Λίστα αντικειμένων

Κλίκ για μεγέθυνση
ABSTRACT
Thyroid hormones (TH) are key regulators of bone health, and TH excess in mice causes high bone turnover–mediated bone loss. However, the underlying molecular mechanisms of TH actions on bone remain poorly defined. Here, we tested the hypothesis whether TH mediate their effects via the pro-osteogenic bone morphogenetic protein (BMP) signaling pathway in vitro and in vivo. Primary murine osteoblasts treated with 3,30
,5-triiodo-L-thyronine (T3) showed an enhanced differentiation potential, which was associated with activated canonical BMP/SMAD signaling reflected by SMAD1/5/8 phosphorylation. Blocking BMP signaling at
the receptor (LDN193189) and ligand level (noggin, anti-BMP2/BMP4 neutralizing antibodies) inhibited T3-induced osteogenic differ-
entiation. In vivo, TH excess over 4 weeks in male C57BL/6JRj mice led to severe trabecular bone loss with a high bone turnover that was completely prevented by treatment with the BMP ligand scavenger ALK3-Fc. Thus, TH activate the canonical BMP pathway in osteoblasts to promote their differentiation and function. Importantly, this study indicates that blocking the BMP pathway may be an effective strategy to treat hyperthyroidism-induced bone loss. © 2020 The Authors. Journal of Bone and Mineral Research published by American Society for Bone and Mineral Research.
INTRODUCTION
Bone is a dynamic organ that underlies constant remodeling. This highly coordinated process is performed by boneresorbing osteoclasts and bone-forming osteoblasts.(1,2) A variety of regulators influence bone cell differentiation, activity, and thereby bone metabolism. Thyroid hormones (TH) are essential determinants of bone mass and quality and contribute
to bone homeostasis and maintenance in adults.(3–5) The prohormone L-thyroxine (T4) and its biologically active form 3,30,5-triiodo-L-thyronine (T3) are produced under the tight control of the hypothalamic-pituitary-thyroid axis. Preclinical and clinical studies showed that untreated hyperthyroidism can result in secondary osteoporosis with increased fracture risk based on an accelerated bone turnover with predominant bone resorption.(6–9) Thus, a balanced TH status is essential for bone
health. Despite these insights and their clinical relevance, the actions of
TH at the molecular level remain largely elusive. Several studies consent that T3 promotes osteogenic differentiation and enhances the synthesis of bone matrix in murine osteoblasts.(10–16) Besides, nuclear TH receptors (TR), TRα1 and TRβ1,(17–20) are expressed in osteoblasts, providing evidence that TH may exert direct effects on osteoblasts. We previously reported that exogenously applied TH enhances bone turnover with an upregulation of both bone formation and predominantly bone resorption, leading to high turnover bone loss in male mice.(9,21) However, in spite of elevated numbers of osteoblasts, Wnt signaling was rather decreased,
showing a high expression of the Wnt inhibitors sclerostin and dickkopf-1 and downregulation of Wnt targets Axin2 and osteoprotegerin in bone tissue.(9) Moreover, deletion of dickkopf-1 did not rescue hyperthyroidism-induced bone loss in mice.(22) Hence, an alternative osteogenesis-promoting pathway such as the bone morphogenetic proteins (BMP) signaling pathway might be activated by TH to promote bone turnover.
BMPs belong to the transforming growth factor β (TGFβ) superfamily and direct the commitment of mesenchymal stemcells (MSC) into osteoprogenitors and further stimulate osteoblastogenesis and bone mineralization.(23–25) Canonical BMP signal transduction is initiated after BMP ligand binding to a heterotetrameric receptor-complex composed of type I and type II BMP transmembrane serine/threonine kinase receptors
(BMPR). Subsequently, constitutive active BMPR type II activates BMPR type I through phosphorylation.(26) BMPR type I phosphorylates receptor activated (R)-SMADs 1, 5, and 8, which can formtrimers with co-receptor (Co)-SMAD 4.(27) After translocation into the nucleus, R-SMAD/Co-SMAD complexes induce transcription of BMP target genes by binding to BMP-responsive elements of their promoter regions and recruiting transcription factors as well as co-activators.(23,25,26) Independent of SMAD proteins,
mitogen-activated protein kinases (MAPK) can be activated through BMPR type I-mediated phosphorylation, leading to genomic and non-genomic pro-osteogenic outcomes.(28–30) Also the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) pathway has been shown to play a role in BMP-mediated signaling in osteoblasts.(31,32) Nonetheless, few studies have investigated the link between TH and BMP signaling in skeletal cells.
In fact, T3 stimulation of chondrocytes increased collagen X mRNA expression mediated by BMP4.(33) Further, Bmp1 expression was upregulated after T3 treatment to stimulate collagen synthesis and cross-linking in osteoblast-like MC3T3 cells via its metalloprotease activity.(12)
To unravel the role of BMP signaling in TH actions on bone, we used inhibitor-based approaches in vitro and in vivo. At the cellular level, T3 promoted osteoblast differentiation and functionality via BMP-SMAD1/5-pathway activation. Blockade of BMP signaling with the selective BMPR type I inhibitor LDN193189 or distinct BMP ligand scavengers reversed T3-mediated effects on osteoblasts. Moreover, treatment of hyperthyroid mice with the BMP ligand trap ALK3-Fc protected against TH excessinduced trabecular bone loss by normalizing bone turnover. Thus, our study reveals that TH exert their osteoinductive effects via the canonical BMP pathway in bone.
Materials and Methods
Animals
All animal procedures were approved by the Institutional Animal Care Committee of the Technische Universität Dresden and the Landesdirektion Dresden. Twelve-week-old male and female C57BL/6JRj mice were purchased from Janvier Labs (Saint Berthevin, France). All mice were housed under institutional guidelines with 12-hour light/dark cycles, fed a standard diet ad libitum, and received cardboard houses for enrichment
purposes. Hyperthyroidism was induced as described previously.(21,22) Mice were randomized into two groups either rendered hyperthyroid (Hyper) by adding L-thyroxine (T4, 1.2 μg/mL, Sigma-Aldrich, Munich, Germany) into their drinking water or remained euthyroid (Co) by receiving tap water. All animals had ad libitum access to their respective drinking water. To study the mechanistic link between thyroid hormones and BMP signaling in bone, only male mice (Co, Hyper) received subcutaneous injections of either 5 mg/kg body weight (BW) ALK3-Fc kindly provided by Acceleron Pharma (Boston,MA, USA) or phosphate-buffered saline (PBS) as a vehicle control (n = 5 per category) twice weekly over 4 weeks. After 4 weeks, both male and female mice were euthanized using CO2 and blood was collected via heart puncture. Serum was obtained by centrifugation at 400g for 15 minutes, and bones were collected for analysis. All subsequent analyses were per-
formed in a blinded fashion.
Serum analysis
Serum concentrations of bone turnover markers procollagen type 1 amino-terminal propeptide (P1NP) and tartrate-resistant acid phosphatase (TRAP) were quantified using ELISAs following the manufacturers ́ protocol (IDS, Frankfurt/Main, Germany). Total T4 and T3 serum concentrations were determined using radioimmunoassays (RIA-4524 and RIA-4525 respectively; DRG Instruments, Marburg, Germany) as previously described.(9,21) The lower limit of analysis was 10 nM and 0.22 nM for T4 and T3, respectively. Intra-assay coefficients of variations were below
7.5%.
Analysis of bone mass and microarchitecture
The distal femur and fourth lumbar vertebra (L4) were analyzed postmortem using a micro-CT (vivaCT 40, Scanco Medical, Brüttisellen, Switzerland) with an X-ray energy of 70 kVp and isotropic voxel size of 10.5 μm (114 mA, 200 msec integration time). All trabecular (Tb) and cortical (Ct) bone parameters are based on calculations including 100 scan slices using standard protocols from Scanco Medical. Trabecular bone of the femora was analyzed in the metaphyseal region extending away from the growth plate, while cortical bone analysis was performed in the
diaphyseal region midway between femoral head and distal condyles. Trabecular bone of L4 was evaluated around the center.
Parameters such as bone volume/total volume (BV/TV), trabecular number (Tb.N), trabecular separation (Tb.Sp), and thickness (Tb.Th) were assessed. Results are presented according to ASBMR guidelines.(34)
Bone histology and histomorphometry Dynamic bone histomorphometry was performed as described previously.(35) Five and 2 days before euthanization, mice received intraperitoneal injections with the fluorochrome calcein (20 mg/kg BW; Sigma-Aldrich, Munich, Germany) that incorporates into newly formed bone. The third lumbar vertebrae were
fixated in 4% PBS-buffered paraformaldehyde for 48 hours and dehydrated via ascending ethanol series. Then, bones were embedded in methyl methacrylate (Technovit 9100, Heraeus Kulzer, Hanau, Germany) and cut into 7-μm sagittal sections for calcein label quantification of the trabecular bone. The mineralized surface/bone surface (MS/BS), the mineral apposition rate (MAR), and the bone formation rate/bone surface (BFR/BS) were quantified in an area of 1.44 mm2 in the center of vertebrae
according to international established protocols(36) using fluorescence microscopy (Microscope Axio Imager M1, Carl Zeiss Jena, Jena, Germany) and the Osteomeasure software (OsteoMetrics, Atlanta, GA, USA). Representative photos were taken using Axio Vision 3.1 Software (Carl Zeiss Jena). Additionally, tartrate-resistant acid phosphatase (TRAP) staining was performed on 2-μm-thick paraffin sagittal sections of the fourth lumbar vertebra, which were prior fixed in 4% PBS-buffered para-
formaldehyde, decalcified in Osteosoft (Merck, Darmstadt, Germany) for 7 days, and dehydrated by ascending ethanol series.
Using Osteomeasure software, osteoclasts as well as osteoblasts were quantified in an area of 0.90 mm2 in the center of vertebrae. Representative photos were taken using Microscope Axio Imager M1 (Carl Zeiss Jena) and CellSens Entry Software Version 1.5 (Olympus Corp, Shinjuku, Japan). Terminology and quantification procedure were conducted according to guidelines of the Nomenclature Committee of the ASBMR.(37)
Biomechanical testing
Before 3-point bending test (Zwick Roell, Ulm, Germany), femurs were rehydrated in PBS overnight. The rehydrated femur was placed onto two supports with an intermediate distance of 6 mm. Mechanical force is applied vertically onto the center of the femoral midshaft. Analysis was initiated after reaching a preload of 1 N and subsequently performed with a load rate of 0.05 mm/s until failure. Results were evaluated using testXpert II - V3.7 software (Zwick Roell).
Cell culture and functional tests of primary murine osteoblasts
Hind legs of wild-type C57BL/6JRj mice were harvested and mesenchymal stromal cells were acquired by flushing the bone marrow. Cells were cultured until 80% confluence in growth medium (DMEM, 10% FCS, 1% P/S) in a humidified atmosphere of 95% air and 5% CO2 at 37C and primary osteoblasts were obtained by adding an osteogenic cocktail to the growth medium that contains 100 μM ascorbate phosphate and 5 mM β-glycerol phosphate (both from Sigma-Aldrich). To assess mineralization
capacity, primary osteoblasts were differentiated and simultaneously treated with indicated concentrations of 3,30,5-triiodo-L-thyronine (T3, 10 to 1000 nM, Sigma-Aldrich), LDN193189 (LDN, 1 to 100 nM, Sigma-Aldrich), or noggin (Nog, 10 to 100 ng/mL, PeproTech, London, UK) over 10 days. Briefly, cells were fixated in 10% paraformaldehyde for 15 minutes and stained with 1% Alizarin red S solution (pH 5.5, Sigma-Aldrich) for 30 minutes at room temperature. Excessive staining was
removed by repeated washing steps with distilled water. Incorporated calcium was dissolved using cetylpyridinium chloride (Sigma-Aldrich) and quantified by its photometric measurement at a wavelength of 540 nm.
Alkaline phosphatase (ALP) activity was evaluated as a marker of osteogenic differentiation and function. To this end, primary osteoblasts were differentiated for 7 days, starved overnight (DMEM, 1% FCS, 1% P/S), and treated over 48 hours with indicated concentrations of T3, LDN193189, or noggin. After washing steps with PBS, cells were scraped in ALP lysis buffer (10 mM TrisHCl pH 8.0, 1 mM MgCl2, and 0.5% Triton X-100) for isolation of total proteins. Subsequently, cell lysates were processed through a 24-gauge needle and centrifuged at 25,000g for 30 minutes at 4C. Ten microliters of diluted supernatants (1:5 in distilled
water) were mixed with 90 μL ALP substrate buffer (100 mM diethanolamine, 150 mM NaCl, 2 mM MgCl2, and 2.5 g/mL p-nitrophenylphosphate) and incubated for 30 minutes at 37C. Color change was observed given the hydrolysis of p-nitrophenylphosphate by enzymatic activity of ALP. Absorbance was assessed at 410 nm using FluoStar Omega (BMG Labtech, Offenburg, Germany). Results were normalized to the total
protein content quantified with Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Schwerte, Germany) according to the manufacturer’s protocol. RNA isolation, reverse transcription, and quantitative real-
time PCR. After 7 days of differentiation, primary osteoblasts were starved
overnight and then treated with indicated concentrations of T3, LDN193189, noggin, or recombinant anti-BMP2 or anti-BMP4 neutralizing antibodies for 48 hours (Supplemental Table S2).
Total RNA was extracted with the High Pure RNA Isolation Kit
(Roche, Mannheim, Germany) according to the manufacturer’s
protocol and quantified using the Nanodrop spectrophotometer
(Peqlab, Erlangen, Germany). RNA (500 ng) was used for reverse transcription using Superscript II (Invitrogen, Darmstadt, Germany) and subsequently analyzed by SYBR green-based real-time PCRs using standard protocol (Applied Biosystems Inc., Carlsbad, CA, USA). PCR conditions were: 50C for 5 minutes and 95C for 10 minutes followed by 40 cycles with 95C for 15 seconds and 60C for 1 minute. Primer sequences for mice are listed in Supplemental Table S1. Melting curves were obtained by the following scheme: 95C for 15 seconds, 60C for 1 minute, and 95C for 30 seconds. Results were calculated according to the ΔΔCT method and are shown in x-fold increase normalized to β-actin mRNA levels.
Western blot analysis
Primary osteoblasts were differentiated over 7 days, starved overnight, and treated with 100 nM T3 for 20 minutes, 40 minutes, and 24 hours. With regard to inhibitor studies, cells were treated with 100 nM T3 and additionally 100 nM LDN193189 over 40 minutes. Subsequently, cells were lysed in lysis buffer containing 20 mM Tris/HCl pH 7.4, 1% SDS, a prote-
ase inhibitor cocktail (complete mini, Roche), and a phosphatase inhibitor cocktail (PhosSTOP, Roche). Samples were centrifuged at 25,000g at 4C for 20 minutes and total protein from supernatants was quantified using the BCA method. For electrophoresis, 20 μg protein was loaded on a 10% SDS-PAGE and subsequently transferred onto a 0.2 μm nitrocellulose membrane (Whatman). After 1 hour of blocking in 5% BSA in Tris-buffered saline with 1% Tween-20 (TBS-T), membranes were incubated overnight at 4C with primary antibodies listed in Supplemental Table S3. GAPDH was used as a loading control. Membranes were washed and incubated with the appropriate anti-rabbit or anti-mouse HRP conjugated secondary antibodies as listed in Supplemental Table S3. After washing and incubating the membranes with an ECL substrate (Pierce, Thermo Fisher Scientific), protein bands were visualized by MF-ChemiBIS 3.2 bioimaging system (Biostep, Jahnsdorf, Germany). The densitometric analysis of
the signal intensities was quantified using ImageJ 1.44 and normalized to GAPDH as the housekeeping protein and the corresponding unphosphorylated form of the protein of interest.
Statistical analysis
Data are shown as means standard deviation. Statistical analysis comparing two groups are based on a two-sided Student’s t test. One-way analysis of variance (ANOVA) or two-way ANOVA were performed for in vitro and in vivo experiments with more than two groups followed by Bonferroni’s multiple comparison post hoc test using GraphPad Prism 7.0 (GraphPad, La Jolla, CA, USA). Values of p < 0.05 were considered statistically significant.
Results
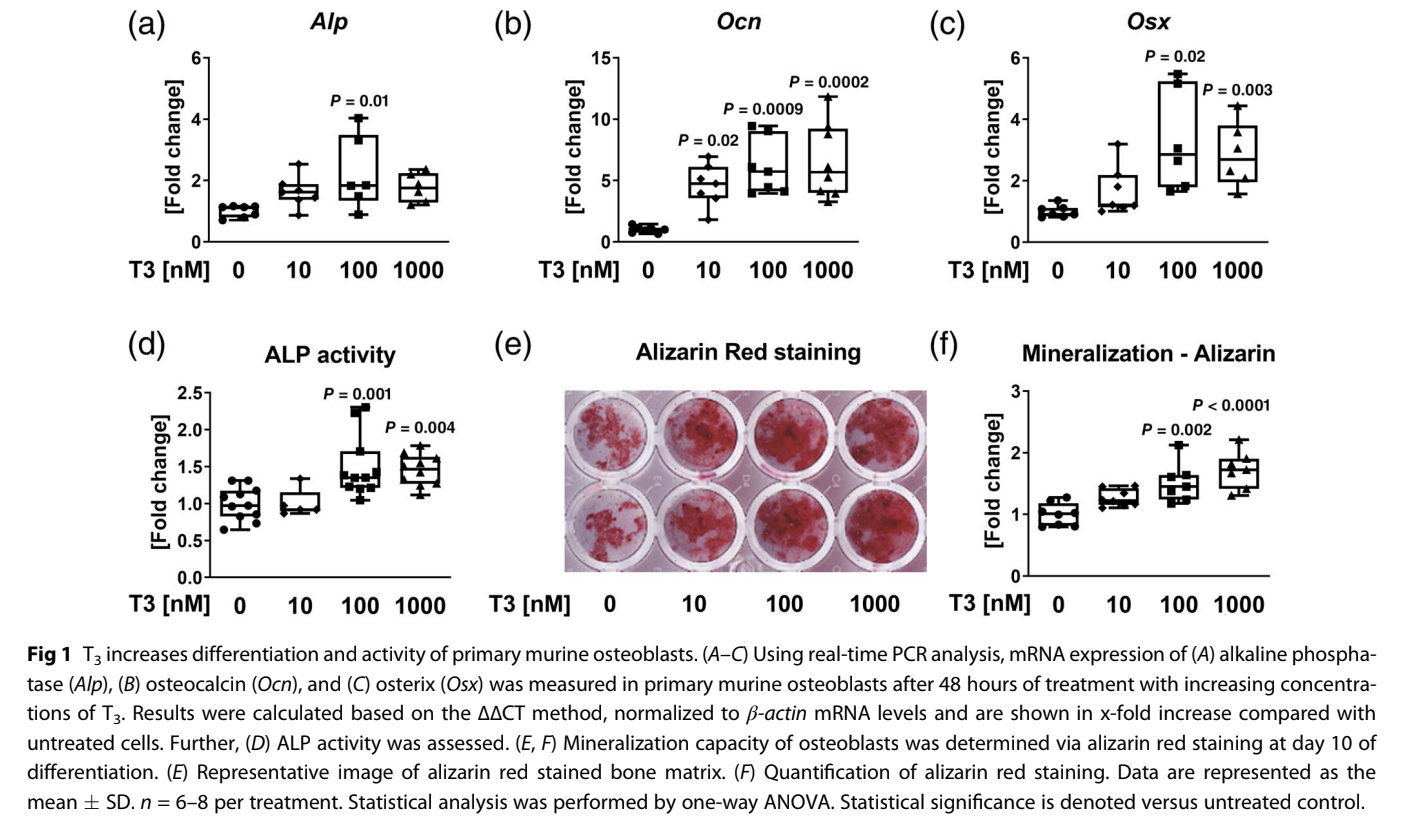
T3 enhances osteoblast differentiation and activity and activates canonical BMP signaling Treatment of primary murine osteoblasts with T3 led to
enhanced expression of differentiation marker genes including alkaline phosphatase (Alp; 100 nM T3: 2.2-fold), osteocalcin(Ocn; 100 nM T3: 6.6-fold), and osterix (Osx; 100 nM T3: 2.7-fold) in a dose-dependent manner (Fig. 1A–C). Moreover, osteoblast function was increased with T3 as shown by elevated ALP activity (100 nM T3: 1.6-fold) and mineralization capacity (100 nM T3: 1.5-fold) (Fig. 1D–F). Further, prominent BMP target genes
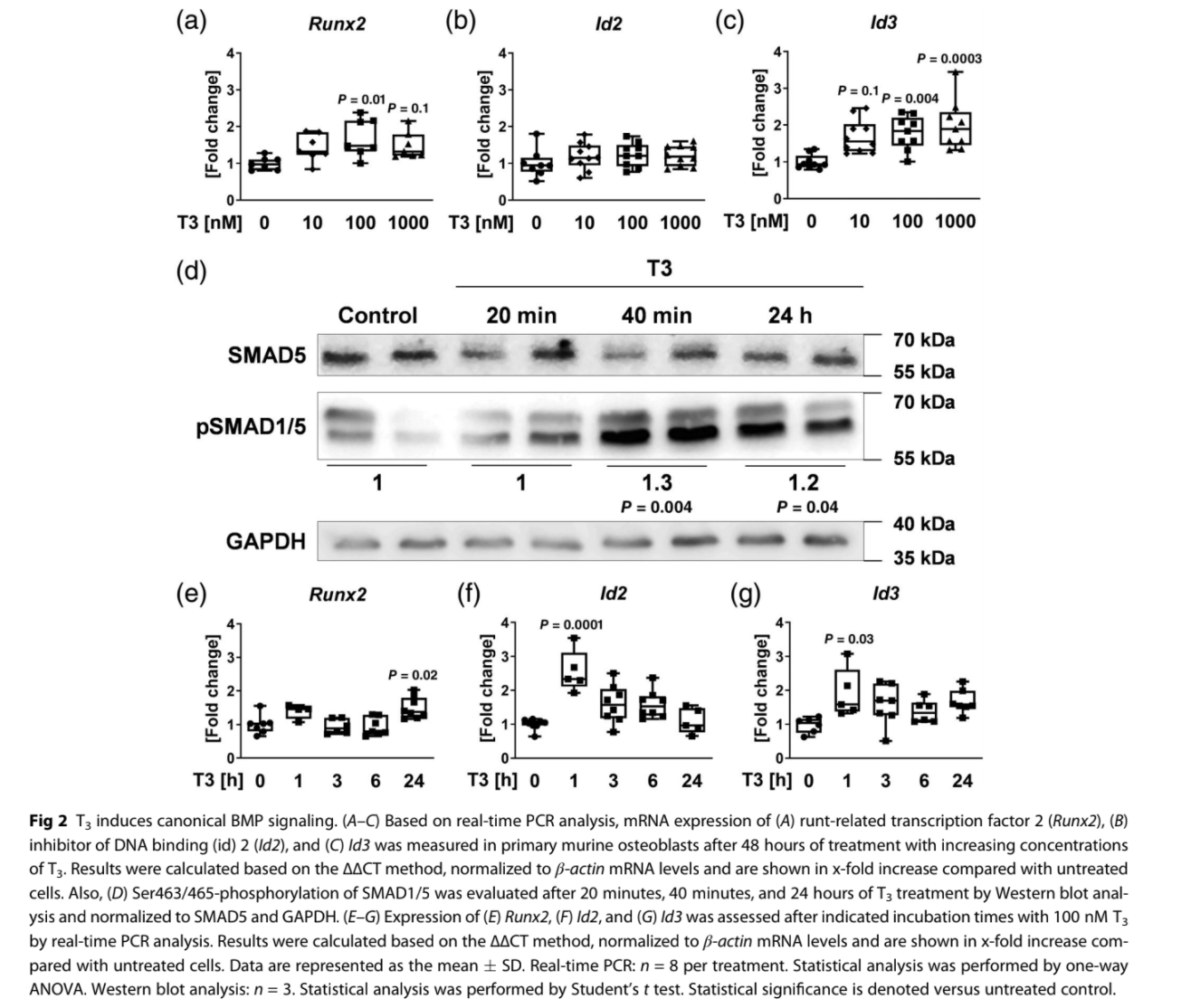
Runt-related transcription factor 2 (Runx2; 100 nM T3: 2-fold) and Inhibitor of DNA binding 3 (Id3; 100 nM T3: 2-fold) were transcriptionally upregulated after 48 hours of T3 treatment (Fig. 2A–C). Western blot analysis revealed canonical BMP pathway activation by enhanced SMAD1/5 phosphorylation after 40 minutes stimulation with 100 nM T3 indicating an immediate response at the protein level (Fig. 2D). SMAD-independent BMP
signaling was not activated as demonstrated by unaltered (pERK1/2; pP38) or even reduced phosphorylation (pAKT) of respective enzymes (Supplemental Fig. S1). Subsequently, mRNA levels of BMP target genes were assessed in T3-treated osteoblasts with regard to different treatment durations (1, 3, 6, 24 hours) uncovering peak expressions of Id2 and Id3 after 1 hour (2.5-fold/2.6-fold) (Fig. 2E–G). In conclusion, T3 directly
activates canonical BMP signaling via SMAD1/5 phosphorylation
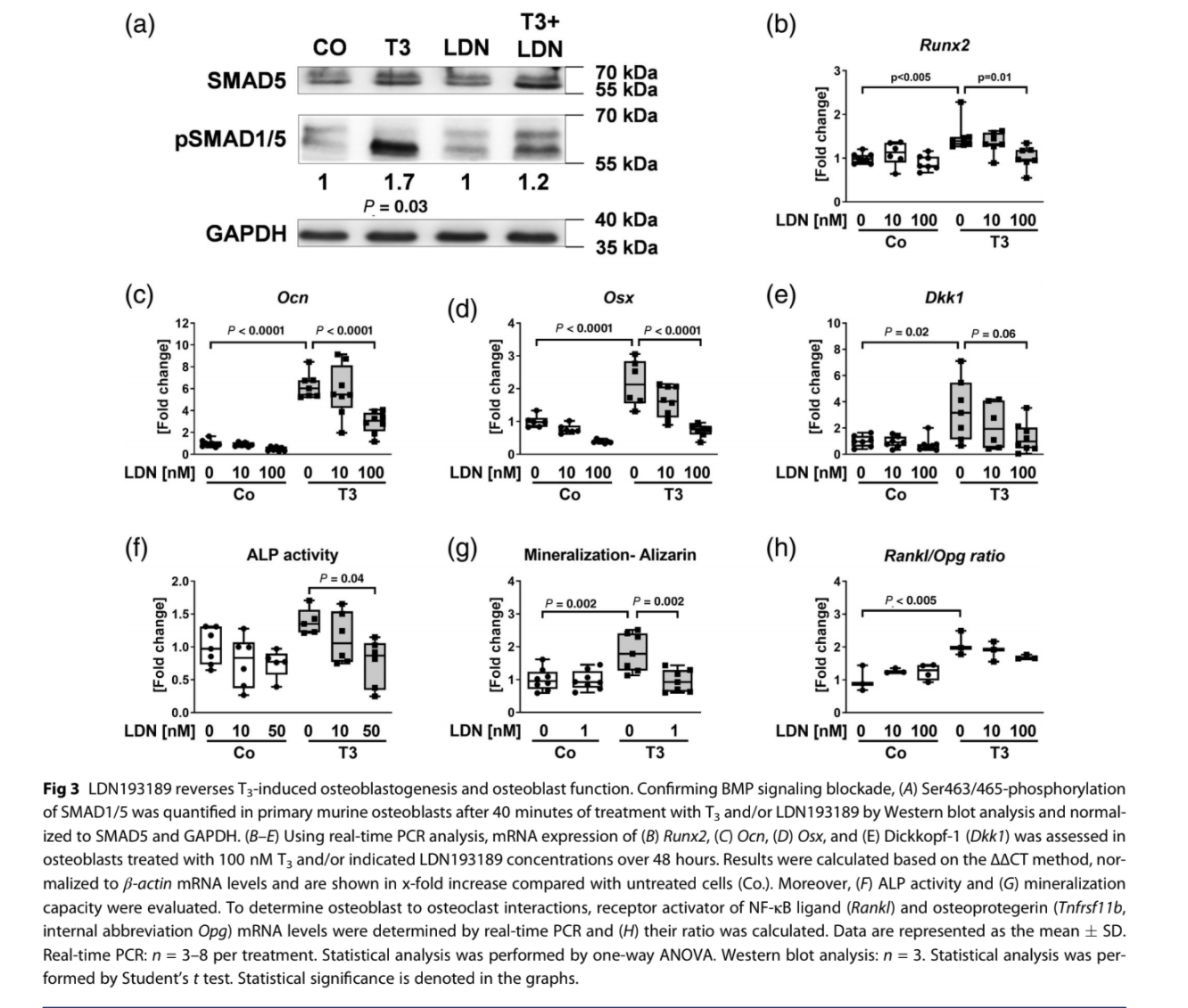
with immediate transcriptional outcome. Blockade of BMP signaling by LDN193189 reverses T3- mediated effects in osteoblasts. To investigate whether T3-promoted osteoblastogenesis is mediated by BMP signaling, we inhibited its activation by specifically blocking BMPR type I members ALK2 and ALK3 using the small molecule LDN193189.(38,39) First, we confirmed signaling inhibition in T3-treated murine osteoblasts with LDN193189 at the protein level as indicated by a reduced SMAD1/5 phosphorylation status (Fig. 3A). Also, T3-induced expression of BMP target Runx2 as well as of osteogenic markers Ocn and Osx was downregulated after BMP blockade (Fig. 3B–D). In line with findings of
Kamiya and colleagues on Wnt inhibitor dickkopf-1 (Dkk1) expression being positively regulated by BMP signaling,(40) Dkk1 was upregulated by T3 and subsequently downregulated by adding LDN193189 (Fig. 3E). T3-mediated repression of Wnt target genes Axin2 and osteoprotegerin (Tnfrsf11b, internal abbreviation Opg), however, remained unchanged by
LDN193189 treatment (Fig. S2A, B). ALP activity and mineralization capacity of T3-stimulated osteoblasts were normalized through ALK2/ALK3 inhibition (Fig. 3F, G). As osteoblasts can regulate osteoclastogenesis by secretion of receptor activator of NF-κB ligand (RANKL) versus OPG, we further assessed the Rankl/Opg ratio. T3 treatment of osteoblasts resulted in an increased Rankl/Opg ratio likely benefiting osteoclast formation,
while LDN193189 normalized the ratio (Fig. 3H and Supplemental Fig. S2C).
BMP ligand scavenger noggin mitigates T3-induced osteogenic response
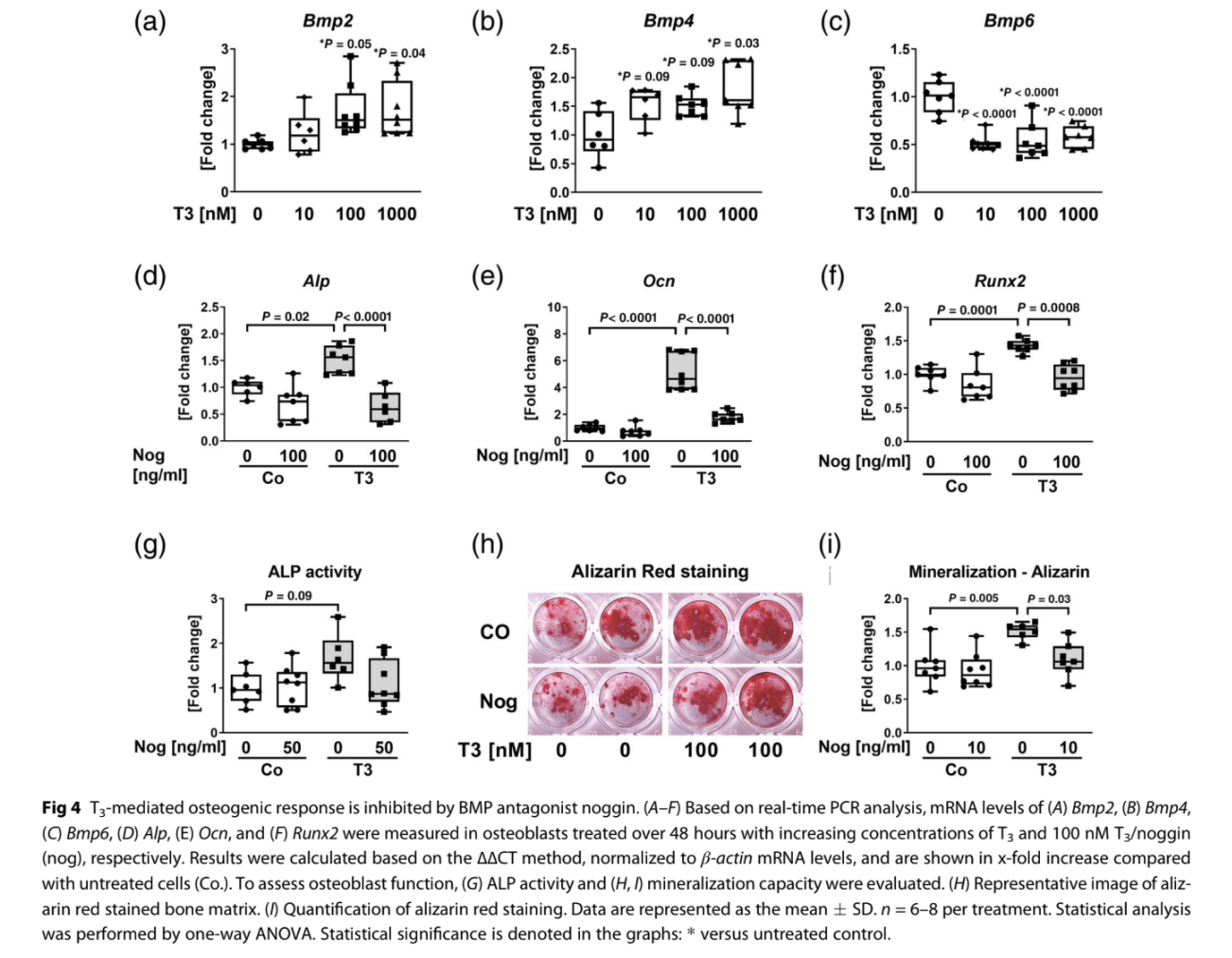
BMP signaling is activated upon BMP ligand binding to respective receptors. So far, BMP2, BMP4, BMP6, BMP7, and BMP9 were described as osteoinductive BMP ligands favoring commitment of MSC to the osteoblast lineage and further stimulating osteoblast differentiation and bone formation.(23–25) T3 increased expression of Bmp2 (100 nM T3: 1.7-fold) and Bmp4 (100 nM T3: 1.5-fold), whereas it reduced Bmp6 mRNA levels (100 nM T3:−46.2%) ex vivo (Fig. 4A–C). Expression of Bmp7 and Bmp9 was below the real-time PCR detection range. To test whether BMP ligands are crucially involved in T3-induced osteoblast activity, we used noggin, which sequesters specifically BMP2 and BMP4. Indeed, noggin normalized T3-activated expression of osteoblastic genes and osteoblast function (Fig. 4D–I). In addition, treatment of hyperthyroid osteoblasts with either specific antiBMP2 or anti-BMP4 neutralizing antibodies reversed T3-induced
expression of osteoblast marker genes (Supplemental Fig. Supplemental Fig. S3A, B) and BMP target genes (Supplemental Fig. Supplemental Fig. S3C, D). Thus, BMP signaling blockade at the receptor but also extracellular ligand level reversed T3-mediated effects on osteoblast differentiation and activity. BMP ligand trap ALK3-Fc protects against
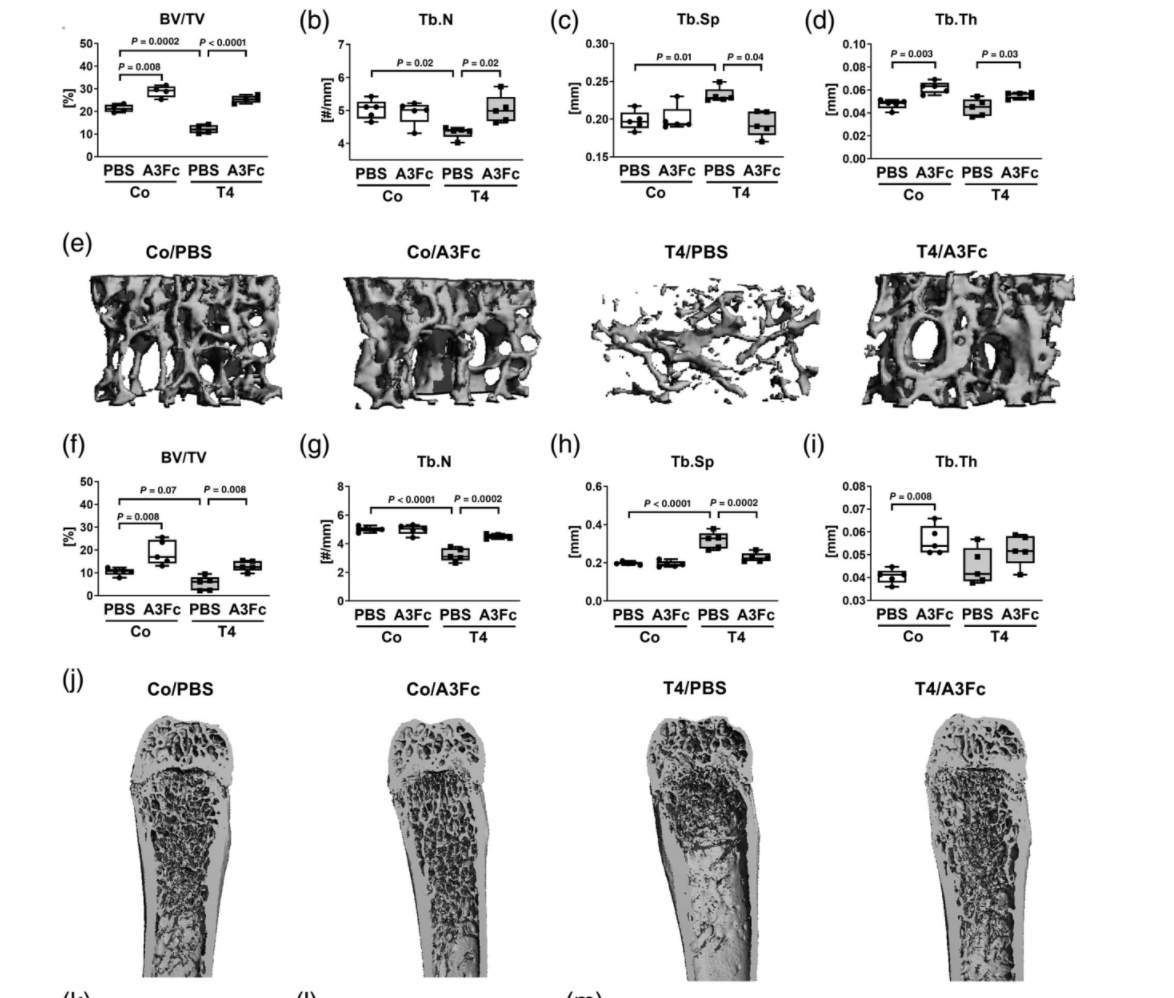
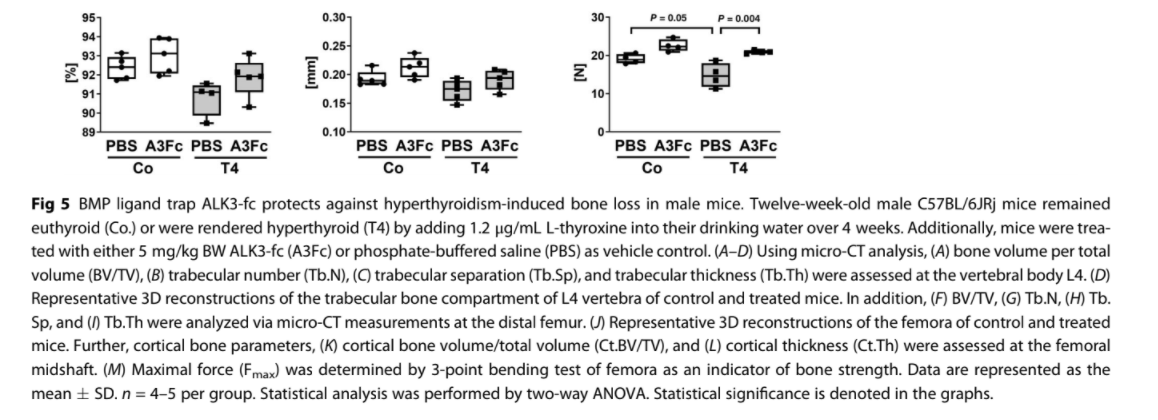
hyperthyroidism-induced bone loss in male mice Hyperthyroidism leads to high bone turnover–induced bone loss due to predominant bone resorption.(9,21) Thus, we tested whether blockade of BMP signaling by scavenging BMP ligands may counteract the accelerated bone turnover and subsequent bone loss. To that end, we treated male hyperthyroid mice with ALK3-Fc, which was reported to sequester ALK3-targeting ligands such as BMP2 and BMP4. Treatment with ALK3-Fc was well-tolerated and no adverse effects were detected. Efficient induction of hyperthyroidism was shown by significant increases of total T3 and T4 serum concentrations in male T4-treated animals (Supplemental Fig. Supplemental Fig. S4). First, using micro-CT measurements, we confirmed trabecular bone loss at the L4 vertebra (−42.9%) and femora (−48.9%) of male mice caused by TH excess (Fig. 5A–J), while cortical bone volume
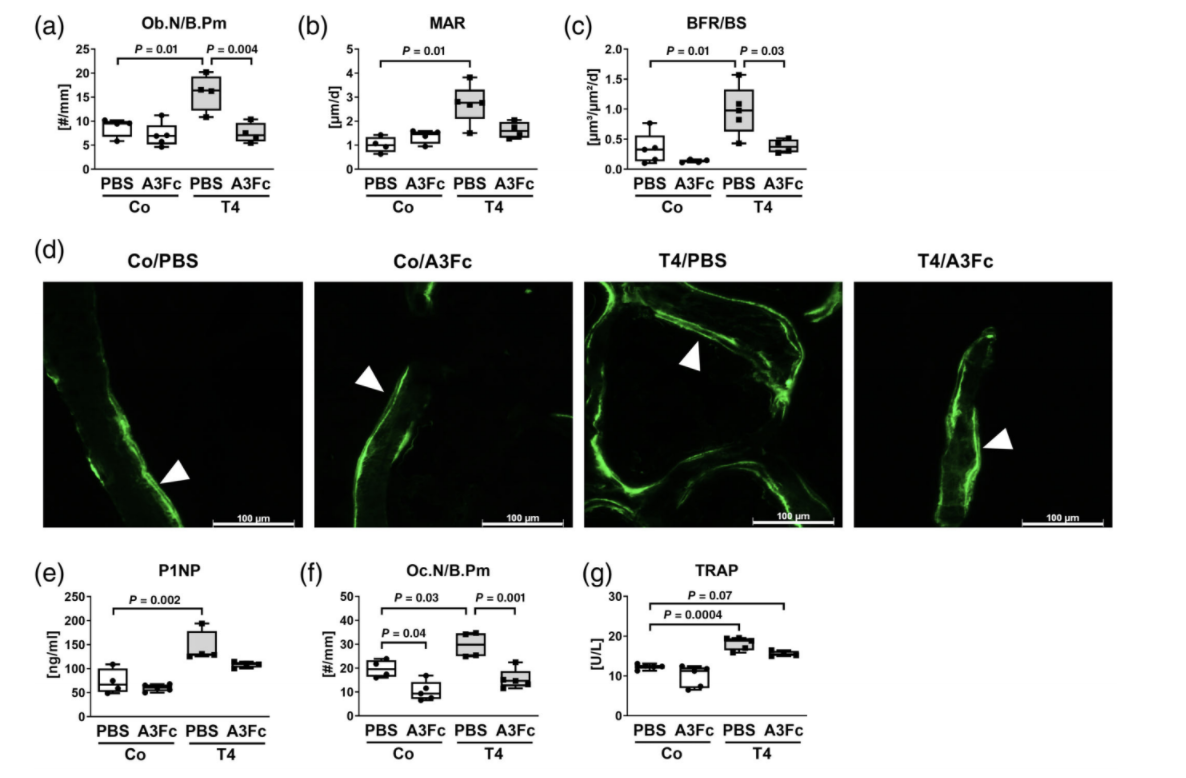
remained unchanged (Fig. 5K, L). Bone changes due to hyperthyroidism were observed in female mice as well (Supplemental Fig. S5). Moreover, bone microstructure analysis revealed a higher bone mass of male hyperthyroid mice treated with ALK3-Fc at the L4 and femur (2.1-fold/2.4-fold versus PBS-treated hyperthyroid mice) and an elevated trabecular number (1.2-fold/1.4-fold versus PBS-treated hyperthyroid mice), whereas trabecular separation was reduced (−16.4%/−28.2% versus PBS-treated hyperthyroid mice), respectively (Fig. 5A–J). ALK3-Fc treatment of euthyroid mice enhanced bone volume (1.4-fold/1.8-fold) and trabecular thickness (1.4-fold/1.4-fold), whereas trabecular number and separation were not altered (Fig. 5A–C). With regard to bone biomechanics, we observed low bone strength in hyperthyroid mice, which was improved
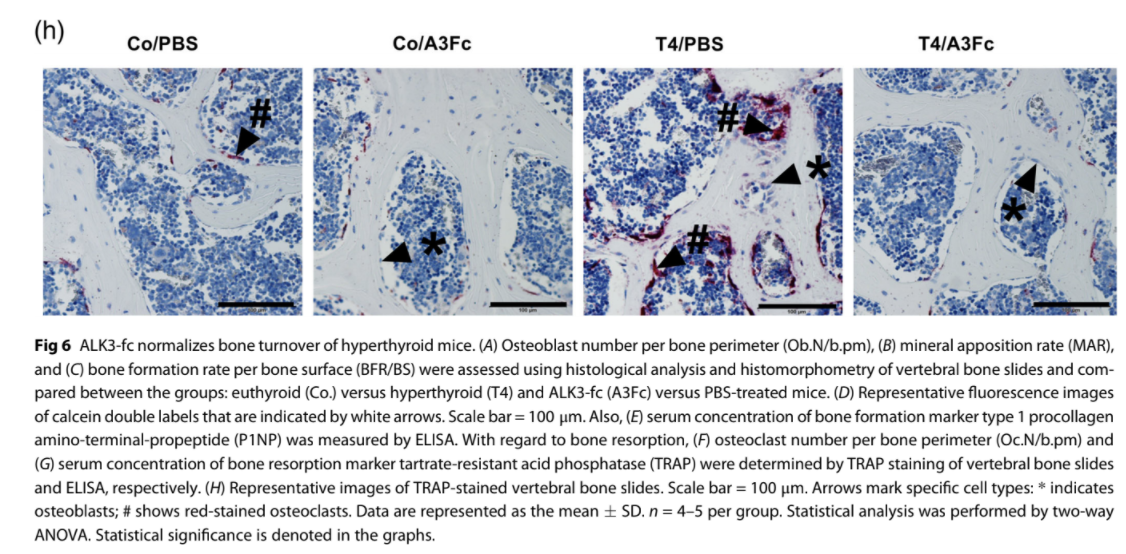
with ALK3-Fc (Fig. 5M). To assess bone formation, histological and bone histomorphometric analyses of vertebral bone slides were performed,
demonstrating augmented osteoblast numbers (1.8-fold), bone formation rate (2.9-fold), and mineral apposition rate (2.7-fold) in hyperthyroid mice (Fig. 6A–D). Inhibition of BMP signaling reversed the enhancement of bone formation parameters as well as the increase in P1NP serum concentrations (Fig. 6A–E). With regard to bone resorption, elevated osteoclast numbers due to hyperthyroidism (1.5-fold) were reduced by ALK3-Fc treatment (−48.3% versus PBS-treated, hyperthyroid mice) at the spine (Fig. 6F). ALK3-Fc also reduced osteoclast numbers in euthyroid
mice (−47.5% versus PBS-treated, euthyroid mice) (Fig. 6F, H). Further, serum concentration of the bone resorption marker TRAP was decreased in hyperthyroid mice with ALK3-Fc treatment (Fig. 6G). In summary, BMP ligand sequestration by ALK3-Fc protected against hyperthyroidism-induced bone loss likely due to normalization of bone formation as well as bone resorption.
Discussion
Bone homeostasis and skeletal health rely on a balanced TH status. In fact, TH excess in mice and humans leads to high bone turnover state with subsequent bone loss due to predominant bone resorption.(6–9,21) Hence, bone-forming osteoblasts as well as bone-resorbing osteoclasts are targeted by TH.(3,41–45) Similar to previous studies,(10–14) our results demonstrate that T3 augments osteoblast differentiation and function in vitro and in vivo. Moreover, we show that the pro-osteogenic effects of
T3 are mediated via activating canonical BMP signaling.
Our study reveals that T3 activates canonical but not noncanonical BMP signaling via rapid and more prolonged effects.
In line with in vitro studies that used BMP7, BMP4, or BMP2,(46–48) T3 induced BMP target gene expression within 1 hour. How this is achieved mechanistically is currently not known, but different scenarios can be envisaged. Considering that BMP must first bind to the BMPR and induce downstream SMAD signaling, the immediate effects of T3 on Id gene expression may indicate cooperative effects of SMADs with either liganded or unliganded TR directly in the nucleus. In fact, Alonso-Merino and colleagues reported on unoccupied TRs associating with SMAD4 and SMAD3 to enhance TGF-β signaling by increasing the activity or stability of transcriptional complexes. Interestingly, T3-binding obstructs this interaction, thereby inhibiting signaling transduction.(49) Finally, they showed that both TRα or TRβ were mediating these effects.(49) Whether TRα or TRβ are involved in BMP signaling in bone remains to be identified in future studies. Besides its effects on transcription, T3 enhanced SMAD1/5 phosphorylation after 40 minutes but not non-canonical SMAD-independent pathways. However, T3 was shown to induce PI3K-AKT signaling after 2 to 5 minutes but not 20 to 30 minutes in a previous study,(50) indicating specific time frames of distinct pathway activation. Also, p38 and ERK1/2-mediated signaling was activated with T3 in MC3T3-E1 cells after 1 hour and 3 hours stimulating osteocalcin synthesis and ALP activity, respectively.(51,52) Additional investigations should focus on time-dependent effects of T3 on SMAD dependent versus SMAD-independent pathway activation to resolve these discrepancies.
At the extracellular level, BMP signaling is tightly regulated via numerous ligands, agonists as well as antagonists. The mRNA levels of BMP ligands Bmp2 and Bmp4 but not Bmp6 were elevated with T3 after 48 hours. Noggin is a key inhibitor of BMP signaling by sequestering specifically BMP2 and BMP4 ligands.
Interestingly, noggin overexpression(53) and osteoblast-specific noggin inactivation(54) both lead to osteopenia, characterized by low bone mineral density and reduced bone formation. These studies suggest that either high BMP signaling activation can exhibit adverse effects on bone mass or that noggin can act BMP-independently on bone. Subsequently, anti-BMP2 and anti-BMP4 antibodies were used to selectively block these
ligands. Interestingly, both treatments were sufficient to abrogate T3-enhanced BMP signaling activation and osteoblast activity. Thus, T3-mediated induction of BMP2 and BMP4 may represent an autocrine feed-forward loop that is involved in the pro-osteogenic effects of T3. However, not only sequestering BMP ligands but also blocking BMPR type I-mediated signaling transduction with LDN193189 reversed the positive effects of
T3 on osteoblast differentiation. T3-dependent effects on bone were also mitigated by blocking BMP signaling in vivo. Using our established protocol, we are able to induce hyperthyroidism-induced bone loss in both
male and female mice. However, we included only male mice
in this initial mechanistic investigation, because several studies
implicated a close cross-talk between BMP and estrogen recep-
tor signaling in osteoblasts/bone that could introduce unex-
pected variables in our experiments.(55,56) To test whether BMP signaling activation mediates thyroid hormone actions in bone, we treated male mice with the BMP antagonist ALK3-Fc that has been previously shown to increase bone mass in healthy mice and to prevent estrogen deficiency–induced osteoporosis.(57) Mechanistically, ALK3-Fc promotes bone formation and reduces bone resorption.(57) In our study, however, ALK3-Fc mitigated hyperthyroidism-induced bone loss by normalizing bone turnover, ie, reducing bone formation and bone resorption parameters to control levels. This is in line with studies on Alk3 knockout mice, which show increased bone mass with low bone turnover, suggesting that the reduction of bone resorption is more severe than the reduction of bone formation.(58–61) The suppression of osteoclastogenesis in mice lacking Alk3 specifically in osteoblasts or osteocytes seems to be mediated via a low RANKL/OPG ratio in Alk3 deficient osteogenic cells.(58) Similarly, we demonstrated a normalization of the Rankl/Opg ratio by BMP inhibition ex vivo that might contribute to reduced osteoclast numbers. In addition,
osteoclasts might be directly influenced by BMP signaling. Genetic and pharmacological blockade of BMP signaling has been shown to affect osteoclasts in vitro and in vivo, especially restricting osteoclastic formation and function.(38,62,63) Further, exogenously applied BMP2 can promote RANKL-mediated osteoclastogenesis and osteoclast activity, implying that BMP locally secreted by osteoblasts might trigger those effects in a
similar manner.(64) Vice versa, BMP signaling in osteoclasts also appears to alter osteoblast function as inhibited SMAD1/5 signaling in osteoclasts has been shown to increase osteoblast activity via osteoclast-to-osteoblast coupling factors such as WNT1, gap junction alpha-1 protein, and sphingosine kinase 1.(65)
Finally, recent reports indicate tight connections between BMP and Wnt signaling.(40,66) in vitro blockade of the BMP pathway reversed T3-stimulated Dkk1 expression but did not alter the expression of Wnt target genes Axin2 nor Opg, suggesting additional mechanisms might obstruct Wnt signaling in osteoblasts.
Previous studies demonstrated that unliganded TRβ binds and stabilizes β-catenin, while T3-binding dissociates their physical connection, leading to β-catenin degradation and subsequent inhibition of canonical Wnt signaling.(67,68) Therefore, canonical Wnt signaling might be regulated independently from BMP signaling in hyperthyroid mice.
Taken together, our study revealed that T3 augments osteoblastogenesis and osteoblast activity via canonical BMP signaling. Moreover, blocking BMP signaling may be an effective strategy to treat hyperthyroidism-induced bone loss.
Disclosures
MR reports honoraria for lectures from Amgen. LCH reports honoraria for advisory boards from Alexion, Amgen, Merck, Radius, Roche, Shire, and UCB to his institution and himself. ET reports honoraria for lectures from Amgen and educational grants from UCB and Shire. RK is employed by Acceleron Pharma, Inc. JK reports honoraria for lectures from Berlin-Chemie, Ipsen, and Sanofi/Genzyme to his institution and himself. The remaining authors have nothing to disclose.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft Schwerpunktprogramm 1629 ThyroidTransAct (to LCH and MR). We thank Ina Gloe, Tina Dybek, Maria Heier, Sandra Hippauf, and Eva Schubert for excellent technical assistance. Authors’ roles: FL and MR designed the experiments. FL, HW, ET, and MR performed all mouse and in vitro experiments and analyzed the data. All authors contributed to data discussion and interpretation. FL and MR drafted the manuscript. All authors critically read, revised, and approved the final version of the
manuscript.
References
1. Crockett JC, Rogers MJ, Coxon FP. Bone remodelling at a glance. J Cell
Sci. 2011;124:991–8.
2. Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone
remodeling. J Biol Chem. 2010;285:25103–8.
3. Bassett JHD, Williams GR. Role of thyroid hormones in skeletal devel-
opment and bone maintenance. Endocr Rev. 2016;37:135–87.
4. Waung JA, Bassett JHD, Williams GR. Thyroid hormone metabolism in
skeletal development and adult bone maintenance. Trends Endocri-
nol Metab. 2012;23:155–62.
5. Lazarus JH, Obuobie K. Thyroid disorders-an update. Postgrad Med J.
2000;76:529–36.
6. Vestergaard P, Mosekilde L. Hyperthyroidism, bone mineral, and frac-
ture risk—a meta-analysis. Thyroid. 2003;13:585–93.
7. Vestergaard P, Rejnmark L, Mosekilde L. Influence of hyper- and
hypothyroidism, and the effects of treatment with antithyroid drugs
and levothyroxine on fracture risk. Calcif Tissue Int. 2005;77:139–44.
8. Nicholls JJ, Brassill MJ, Williams GR. The skeletal consequences of thy-
rotoxicosis. J Endocrinol. 2012;213:209–21.
9. Tsourdi E, Rijntjes E, Köhrle J. Hyperthyroidism and hypothyroidism in
male mice and their effects on bone mass, bone turnover, and the
Wnt inhibitors sclerostin and dickkopf-1. Endocrinology. 2015;156:
3517–27.
10. Fratzl-Zelman N, Hörandner H, Luegmayr E. Effects of triiodothyro-
nine on the morphology of cells and matrix, the localization of alka-
line phosphatase, and the frequency of apoptosis in long-term
cultures of MC3T3-E1 cells. Bone. 1997;20:225–36.
11. Klaushofer K, Varga F, Glantschnig H. The regulatory role of thyroid
hormones in bone cell growth and differentiation. J Nutr. 2018;125:
1996S–2003S.
12. Varga F, Rumpler M, Zoehrer R. T3 affects expression of collagen I and
collagen cross-linking in bone cell cultures. Biochem Biophys Res
Commun. 2010;402:180–5.
13. Banovac K, Koren E. Triiodothyronine stimulates the release of
membrane-bound alkaline phosphatase in osteoblastic cells. Calcif
Tissue Int. 2000;67:460–5.
14. Tokuda K, Otsuka T, Adachi S. (−)-epigallocatechin gallate inhibits
thyroid hormone-stimulated osteocalcin synthesis in osteoblasts.
Mol Med Rep. 2011;4:297–300.
15. Cray JJ, Khaksarfard K, Weinberg SM. Effects of thyroxine exposure on
osteogenesis in mouse Calvarial pre-osteoblasts. PLoS One. 2013;8:
e69067.
16. Huang BK, Golden LA, Tarjan G. Insulin-like growth factor I produc-
tion is essential for anabolic effects of thyroid hormone in osteo-
blasts. J Bone Miner Res. 2010;15:188–97.
17. Siddiqi A, Parsons MP, Lewis JL. TR expression and function in human
bone marrow stromal and osteoblast-like cells. J Clin Endocrinol
Metab. 2002;87:906–14.
18. Gruber R, Czerwenka K, Wolf F. Expression of the vitamin D receptor,
of estrogen and thyroid hormone receptor alpha- and beta-isoforms,
and of the androgen receptor in cultures of native mouse bone mar-
row and of stromal/osteoblastic cells. Bone. 1999;24:465–73.
19. Allain TJ, Yen PM, Flanagan AM. The isoform-specific expression of
the tri-iodothyronine receptor in osteoblasts and osteoclasts. Eur J
Clin Invest. 1996;26:418–25.
20. Krieger NS, Stappenbeck TS, Stern PH. Characterization of specific
thyroid hormone receptors in bone. J Bone Miner Res. 1988;3:473–8.
21. Tsourdi E, Lademann F, Ominsky MS. Sclerostin blockade and zole-
dronic acid improve bone mass and strength in male mice with exog-
enous hyperthyroidism. Endocrinology. 2017;158:3765–77.
22. Tsourdi E, Colditz J, Lademann F. The role of Dickkopf-1 in thyroid
hormone-induced changes of bone remodeling in male mice. Endo-
crinology. 2019;160:664–74.
23. Rahman MS, Akhtar N, Jamil HM. TGF-β/BMP signaling and other
molecular events: regulation of osteoblastogenesis and bone forma-
tion. Bone Res. 2015;3:15005.
24. Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein
receptors and signal transduction. J Biochem. 2010;147:35–51.
25. Wu M, Chen G, Li YP. TGF-β and BMP signaling in osteoblast, skeletal
development, and bone formation, homeostasis and disease. Bone
Res. 2016;4:16009.
26. Beederman M, Lamplot JD, Nan G. BMP signaling in mesenchymal
stem cell differentiation and bone formation. J Biomed Sci Eng.
2013;6:32–52.
27. Massague J, Seoane J, Wotton D. Smad transcription factors. Genes
Dev. 2005;19:2783–810.
28. Greenblatt MB, Shim J-H, Zou W. The p38 MAPK pathway is essential
for skeletogenesis and bone homeostasis in mice. J Clin Invest. 2010;
120:2457–73.
29. Ortuño MJ, Ruiz-Gaspà S, Rodríguez-Carballo E. p38 regulates expres-
sion of osteoblast-specific genes by phosphorylation of Osterix. J Biol
Chem. 2010;285:31985–94.
30. Choi YH, Gu Y-M, Oh J-W. Osterix is regulated by Erk1/2 during oste-
oblast differentiation. Biochem Biophys Res Commun. 2011;415:472–8.
31. Ge C, Yang Q, Zhao G. Interactions between extracellular signal-
regulated kinase 1/2 and p38 MAP kinase pathways in the control
of RUNX2 phosphorylation and transcriptional activity. J Bone Miner
Res. 2012;27:538–51.
32. Lauzon M-A, Drevelle O, Daviau A. Effects of BMP-9 and BMP-2 on the
PI3K/Akt pathway in MC3T3-E1 preosteoblasts. Tissue Eng Part A.
2016;22:1075–85.
33. Lassová L, Niu Z, Golden EB. Thyroid hormone treatment of cultured
chondrocytes mimics in vivo stimulation of collagen X mRNA by
increasing BMP 4 expression. J Cell Physiol. 2009;219:595–605.
34. Bouxsein ML, Boyd SK, Christiansen BA. Guidelines for assessment of
bone microstructure in rodents using micro-computed tomography.
J Bone Miner Res. 2010;25:1468–86.
35. Rauner M, Föger-Samwald U, Kurz MF. Cathepsin S controls adipocy-
tic and osteoblastic differentiation, bone turnover, and bone micro-
architecture. Bone. 2014;64:281–7.
36. Rauner M, Franke K, Murray M. Increased EPO levels are associated
with bone loss in mice lacking PHD2 in EPO-producing cells. J Bone
Miner Res. 2016;31:1877–87.
37. Dempster DW, Compston JE, Drezner MK. Standardized nomencla-
ture, symbols, and units for bone histomorphometry: a 2012 update
of the report of the ASBMR Histomorphometry Nomenclature Com-
mittee. J Bone Miner Res. 2013;28:2–17.
38. Boergermann JH, Kopf J, Yu PB. Dorsomorphin and LDN-193189
inhibit BMP-mediated Smad, p38 and Akt signalling in C2C12 cells.
Int J Biochem Cell Biol. 2010;42:1802–7.
39. Yu PB, Deng DY, Lai CS. BMP type I receptor inhibition reduces het-
erotopic ossification. Nat Med. 2008;14:1363–9.
40. Kamiya N, Kobayashi T, Mochida Y. Wnt inhibitors Dkk1 and sost are
downstream targets of BMP signaling through the type IA receptor
(BMPRIA) in osteoblasts. J Bone Miner Res. 2010;25:200–10.
41. Klaushofer K, Hoffmann O, Gleispach H. Bone-resorbing activity of
thyroid hormones is related to prostaglandin production in cultured
neonatal mouse calvaria. J Bone Miner Res. 2009;4:305–12.
42. Mundy GR, Shapiro JL, Bandelin JG. Direct stimulation of bone
resorption by thyroid hormones. J Clin Invest. 1976;58:529–34.
43. Allain TJ, Chambers TJ, Flanagan AM. Tri-iodothyronine stimulates rat
osteoclastic bone resorption by an indirect effect. J Endocrinol. 1992;
133:327–31.
44. Siddiqi A, Burrin JM, Wood DF. Tri-iodothyronine regulates the pro-
duction of interleukin-6 and interleukin-8 in human bone marrow
stromal and osteoblast-like cells. J Endocrinol. 1998;157:453–61.
45. Miura M, Tanaka K, Komatsu Y. A novel interaction between thyroid
hormones and 1,25(OH)2D3 in osteoclast formation. Biochem Bio-
phys Res Commun. 2002;291:987–94.
46. Korchynskyi O, ten Dijke P. Identification and functional characteriza-
tion of distinct critically important bone morphogenetic protein-
specific response elements in the Id1 promoter. J Biol Chem. 2002;
277:4883–91.
47. Hollnagel A, Oehlmann V, Heymer J. Id genes are direct targets of
bone morphogenetic protein induction in embryonic stem cells.
J Biol Chem. 1999;274:19838–45.
48. López-Rovira T, Chalaux E, Massagué J. Direct binding of Smad1 and
Smad4 to two distinct motifs mediates bone morphogenetic protein-
specific transcriptional activation of Id1 gene. J Biol Chem. 2002;277:
3176–85.
49. Alonso-Merino E, Martín Orozco R, Ruíz-Llorente L. Thyroid hormones
inhibit TGF-β signaling and attenuate fibrotic responses. Proc Natl
Acad Sci. 2016;113:E3451–60.
50. Kalyanaraman H, Schwappacher R, Joshua J. Nongenomic thyroid
hormone signaling occurs through a plasma membrane -localized
receptor. Sci Signal. 2014;7:ra48.
51. Ishisaki A, Tokuda H, Yoshida M. Activation of p38 mitogen-activated
protein kinase mediates thyroid hormone-stimulated osteocalcin
synthesis in osteoblasts. Mol Cell Endocrinol. 2004;214:189–95.
52. Kozawa O, Hatakeyama D, Yoshida M. Activation of p44/p42
mitogen-activated protein kinase limits triiodothyronine-stimulated
alkaline phosphatase activity in osteoblasts. Biochem Biophys Res
Commun. 2001;286:1140–3.
53. Devlin RD, Du Z, Pereira RC. Skeletal overexpression of noggin results
in osteopenia and reduced bone formation. Endocrinology. 2003;
144:1972–8.
54. Canalis E, Brunet LJ, Parker K. Conditional inactivation of noggin in
the postnatal skeleton causes osteopenia. Endocrinology. 2012;153:
1616–26.
55. Soldin OP, Mattison DR. Sex differences in pharmacokinetics and
pharmacodynamics. Clin Pharmacokinet. 2009;48:143–57.
56. Salama G, Bett GCL. Sex and gender differences in cardiovascular
physiology–back to basics: sex differences in the mechanisms under-
lying long QT syndrome. Am J Physiol. 2014;307:H640.
57. Baud’huin M, Solban N, Cornwall-Brady M. A soluble bone morpho-
genetic protein type IA receptor increases bone mass and bone strength. Proc Natl Acad Sci. 2012;109:12207–12.
58. Kamiya N, Ye L, Kobayashi T. Disruption of BMP signaling in osteo-
blasts through type IA receptor (BMPRIA) increases bone mass.
J Bone Miner Res. 2008;23:2007–17.
59. Kamiya N, Shuxian L, Yamaguchi R. Targeted disruption of BMP sig-
naling through type IA receptor (BMPR1A) in osteocyte suppresses
SOST and RANKL, leading to dramatic increase in bone mass, bone
mineral density and mechanical strength. Bone. 2016;91:53–63.
60. Zhang Y, McNerny EG, Terajima M. Loss of BMP signaling through
BMPR1A in osteoblasts leads to greater collagen cross-link matura-
tion and material-level mechanical properties in mouse femoral trabecular compartments. Bone. 2016;88:74–84.
61. Shi C, Mandair GS, Zhang H. Bone morphogenetic protein signaling
through ACVR1 and BMPR1A negatively regulates bone mass along
with alterations in bone composition. J Struct Biol. 2018;201:237–46.
62. Okamoto M, Murai J, Imai Y. Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J Bone Miner Res. 2011;26:
2511–22.
63. Tasca A, Stemig M, Broege A. Smad1/5 and Smad4 expression are
important for osteoclast differentiation. J Cell Biochem. 2015;116:
1350–60.
64. Jensen ED, Pham L, Billington CJ. Bone morphogenic protein
2 directly enhances differentiation of murine osteoclast precursors.
J Cell Biochem. 2010;109:672–82.
65. Tasca A, Astleford K, Blixt NC. SMAD1/5 signaling in osteoclasts regu-
lates bone formation via coupling factors. PLoS One. 2018;13:
e0203404.
66. Rauner M, Baschant U, Roetto A. Transferrin receptor 2 controls bone
mass and pathological bone formation via BMP and Wnt signalling.
Nat Metab. 2018;1:111–24.
67. O’Shea PJ, Kim DW, Logan JG. Advanced bone formation in mice with
a dominant-negative mutation in the thyroid hormone receptor β
gene due to activation of Wnt/β-catenin protein signaling. J Biol
Chem. 2012;287:17812–22.
68. Guigon CJ, Zhao L, Lu C. Regulation of beta-catenin by a novel non-
genomic action of thyroid hormone receptor. Mol Cell Biol. 2008;
28:4598–608.
Thyroid hormones (TH) are key regulators of bone health, and TH excess in mice causes high bone turnover–mediated bone loss. However, the underlying molecular mechanisms of TH actions on bone remain poorly defined. Here, we tested the hypothesis whether TH mediate their effects via the pro-osteogenic bone morphogenetic protein (BMP) signaling pathway in vitro and in vivo. Primary murine osteoblasts treated with 3,30
,5-triiodo-L-thyronine (T3) showed an enhanced differentiation potential, which was associated with activated canonical BMP/SMAD signaling reflected by SMAD1/5/8 phosphorylation. Blocking BMP signaling at
the receptor (LDN193189) and ligand level (noggin, anti-BMP2/BMP4 neutralizing antibodies) inhibited T3-induced osteogenic differ-
entiation. In vivo, TH excess over 4 weeks in male C57BL/6JRj mice led to severe trabecular bone loss with a high bone turnover that was completely prevented by treatment with the BMP ligand scavenger ALK3-Fc. Thus, TH activate the canonical BMP pathway in osteoblasts to promote their differentiation and function. Importantly, this study indicates that blocking the BMP pathway may be an effective strategy to treat hyperthyroidism-induced bone loss. © 2020 The Authors. Journal of Bone and Mineral Research published by American Society for Bone and Mineral Research.
INTRODUCTION
Bone is a dynamic organ that underlies constant remodeling. This highly coordinated process is performed by boneresorbing osteoclasts and bone-forming osteoblasts.(1,2) A variety of regulators influence bone cell differentiation, activity, and thereby bone metabolism. Thyroid hormones (TH) are essential determinants of bone mass and quality and contribute
to bone homeostasis and maintenance in adults.(3–5) The prohormone L-thyroxine (T4) and its biologically active form 3,30,5-triiodo-L-thyronine (T3) are produced under the tight control of the hypothalamic-pituitary-thyroid axis. Preclinical and clinical studies showed that untreated hyperthyroidism can result in secondary osteoporosis with increased fracture risk based on an accelerated bone turnover with predominant bone resorption.(6–9) Thus, a balanced TH status is essential for bone
health. Despite these insights and their clinical relevance, the actions of
TH at the molecular level remain largely elusive. Several studies consent that T3 promotes osteogenic differentiation and enhances the synthesis of bone matrix in murine osteoblasts.(10–16) Besides, nuclear TH receptors (TR), TRα1 and TRβ1,(17–20) are expressed in osteoblasts, providing evidence that TH may exert direct effects on osteoblasts. We previously reported that exogenously applied TH enhances bone turnover with an upregulation of both bone formation and predominantly bone resorption, leading to high turnover bone loss in male mice.(9,21) However, in spite of elevated numbers of osteoblasts, Wnt signaling was rather decreased,
showing a high expression of the Wnt inhibitors sclerostin and dickkopf-1 and downregulation of Wnt targets Axin2 and osteoprotegerin in bone tissue.(9) Moreover, deletion of dickkopf-1 did not rescue hyperthyroidism-induced bone loss in mice.(22) Hence, an alternative osteogenesis-promoting pathway such as the bone morphogenetic proteins (BMP) signaling pathway might be activated by TH to promote bone turnover.
BMPs belong to the transforming growth factor β (TGFβ) superfamily and direct the commitment of mesenchymal stemcells (MSC) into osteoprogenitors and further stimulate osteoblastogenesis and bone mineralization.(23–25) Canonical BMP signal transduction is initiated after BMP ligand binding to a heterotetrameric receptor-complex composed of type I and type II BMP transmembrane serine/threonine kinase receptors
(BMPR). Subsequently, constitutive active BMPR type II activates BMPR type I through phosphorylation.(26) BMPR type I phosphorylates receptor activated (R)-SMADs 1, 5, and 8, which can formtrimers with co-receptor (Co)-SMAD 4.(27) After translocation into the nucleus, R-SMAD/Co-SMAD complexes induce transcription of BMP target genes by binding to BMP-responsive elements of their promoter regions and recruiting transcription factors as well as co-activators.(23,25,26) Independent of SMAD proteins,
mitogen-activated protein kinases (MAPK) can be activated through BMPR type I-mediated phosphorylation, leading to genomic and non-genomic pro-osteogenic outcomes.(28–30) Also the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) pathway has been shown to play a role in BMP-mediated signaling in osteoblasts.(31,32) Nonetheless, few studies have investigated the link between TH and BMP signaling in skeletal cells.
In fact, T3 stimulation of chondrocytes increased collagen X mRNA expression mediated by BMP4.(33) Further, Bmp1 expression was upregulated after T3 treatment to stimulate collagen synthesis and cross-linking in osteoblast-like MC3T3 cells via its metalloprotease activity.(12)
To unravel the role of BMP signaling in TH actions on bone, we used inhibitor-based approaches in vitro and in vivo. At the cellular level, T3 promoted osteoblast differentiation and functionality via BMP-SMAD1/5-pathway activation. Blockade of BMP signaling with the selective BMPR type I inhibitor LDN193189 or distinct BMP ligand scavengers reversed T3-mediated effects on osteoblasts. Moreover, treatment of hyperthyroid mice with the BMP ligand trap ALK3-Fc protected against TH excessinduced trabecular bone loss by normalizing bone turnover. Thus, our study reveals that TH exert their osteoinductive effects via the canonical BMP pathway in bone.
Materials and Methods
Animals
All animal procedures were approved by the Institutional Animal Care Committee of the Technische Universität Dresden and the Landesdirektion Dresden. Twelve-week-old male and female C57BL/6JRj mice were purchased from Janvier Labs (Saint Berthevin, France). All mice were housed under institutional guidelines with 12-hour light/dark cycles, fed a standard diet ad libitum, and received cardboard houses for enrichment
purposes. Hyperthyroidism was induced as described previously.(21,22) Mice were randomized into two groups either rendered hyperthyroid (Hyper) by adding L-thyroxine (T4, 1.2 μg/mL, Sigma-Aldrich, Munich, Germany) into their drinking water or remained euthyroid (Co) by receiving tap water. All animals had ad libitum access to their respective drinking water. To study the mechanistic link between thyroid hormones and BMP signaling in bone, only male mice (Co, Hyper) received subcutaneous injections of either 5 mg/kg body weight (BW) ALK3-Fc kindly provided by Acceleron Pharma (Boston,MA, USA) or phosphate-buffered saline (PBS) as a vehicle control (n = 5 per category) twice weekly over 4 weeks. After 4 weeks, both male and female mice were euthanized using CO2 and blood was collected via heart puncture. Serum was obtained by centrifugation at 400g for 15 minutes, and bones were collected for analysis. All subsequent analyses were per-
formed in a blinded fashion.
Serum analysis
Serum concentrations of bone turnover markers procollagen type 1 amino-terminal propeptide (P1NP) and tartrate-resistant acid phosphatase (TRAP) were quantified using ELISAs following the manufacturers ́ protocol (IDS, Frankfurt/Main, Germany). Total T4 and T3 serum concentrations were determined using radioimmunoassays (RIA-4524 and RIA-4525 respectively; DRG Instruments, Marburg, Germany) as previously described.(9,21) The lower limit of analysis was 10 nM and 0.22 nM for T4 and T3, respectively. Intra-assay coefficients of variations were below
7.5%.
Analysis of bone mass and microarchitecture
The distal femur and fourth lumbar vertebra (L4) were analyzed postmortem using a micro-CT (vivaCT 40, Scanco Medical, Brüttisellen, Switzerland) with an X-ray energy of 70 kVp and isotropic voxel size of 10.5 μm (114 mA, 200 msec integration time). All trabecular (Tb) and cortical (Ct) bone parameters are based on calculations including 100 scan slices using standard protocols from Scanco Medical. Trabecular bone of the femora was analyzed in the metaphyseal region extending away from the growth plate, while cortical bone analysis was performed in the
diaphyseal region midway between femoral head and distal condyles. Trabecular bone of L4 was evaluated around the center.
Parameters such as bone volume/total volume (BV/TV), trabecular number (Tb.N), trabecular separation (Tb.Sp), and thickness (Tb.Th) were assessed. Results are presented according to ASBMR guidelines.(34)
Bone histology and histomorphometry Dynamic bone histomorphometry was performed as described previously.(35) Five and 2 days before euthanization, mice received intraperitoneal injections with the fluorochrome calcein (20 mg/kg BW; Sigma-Aldrich, Munich, Germany) that incorporates into newly formed bone. The third lumbar vertebrae were
fixated in 4% PBS-buffered paraformaldehyde for 48 hours and dehydrated via ascending ethanol series. Then, bones were embedded in methyl methacrylate (Technovit 9100, Heraeus Kulzer, Hanau, Germany) and cut into 7-μm sagittal sections for calcein label quantification of the trabecular bone. The mineralized surface/bone surface (MS/BS), the mineral apposition rate (MAR), and the bone formation rate/bone surface (BFR/BS) were quantified in an area of 1.44 mm2 in the center of vertebrae
according to international established protocols(36) using fluorescence microscopy (Microscope Axio Imager M1, Carl Zeiss Jena, Jena, Germany) and the Osteomeasure software (OsteoMetrics, Atlanta, GA, USA). Representative photos were taken using Axio Vision 3.1 Software (Carl Zeiss Jena). Additionally, tartrate-resistant acid phosphatase (TRAP) staining was performed on 2-μm-thick paraffin sagittal sections of the fourth lumbar vertebra, which were prior fixed in 4% PBS-buffered para-
formaldehyde, decalcified in Osteosoft (Merck, Darmstadt, Germany) for 7 days, and dehydrated by ascending ethanol series.
Using Osteomeasure software, osteoclasts as well as osteoblasts were quantified in an area of 0.90 mm2 in the center of vertebrae. Representative photos were taken using Microscope Axio Imager M1 (Carl Zeiss Jena) and CellSens Entry Software Version 1.5 (Olympus Corp, Shinjuku, Japan). Terminology and quantification procedure were conducted according to guidelines of the Nomenclature Committee of the ASBMR.(37)
Biomechanical testing
Before 3-point bending test (Zwick Roell, Ulm, Germany), femurs were rehydrated in PBS overnight. The rehydrated femur was placed onto two supports with an intermediate distance of 6 mm. Mechanical force is applied vertically onto the center of the femoral midshaft. Analysis was initiated after reaching a preload of 1 N and subsequently performed with a load rate of 0.05 mm/s until failure. Results were evaluated using testXpert II - V3.7 software (Zwick Roell).
Cell culture and functional tests of primary murine osteoblasts
Hind legs of wild-type C57BL/6JRj mice were harvested and mesenchymal stromal cells were acquired by flushing the bone marrow. Cells were cultured until 80% confluence in growth medium (DMEM, 10% FCS, 1% P/S) in a humidified atmosphere of 95% air and 5% CO2 at 37C and primary osteoblasts were obtained by adding an osteogenic cocktail to the growth medium that contains 100 μM ascorbate phosphate and 5 mM β-glycerol phosphate (both from Sigma-Aldrich). To assess mineralization
capacity, primary osteoblasts were differentiated and simultaneously treated with indicated concentrations of 3,30,5-triiodo-L-thyronine (T3, 10 to 1000 nM, Sigma-Aldrich), LDN193189 (LDN, 1 to 100 nM, Sigma-Aldrich), or noggin (Nog, 10 to 100 ng/mL, PeproTech, London, UK) over 10 days. Briefly, cells were fixated in 10% paraformaldehyde for 15 minutes and stained with 1% Alizarin red S solution (pH 5.5, Sigma-Aldrich) for 30 minutes at room temperature. Excessive staining was
removed by repeated washing steps with distilled water. Incorporated calcium was dissolved using cetylpyridinium chloride (Sigma-Aldrich) and quantified by its photometric measurement at a wavelength of 540 nm.
Alkaline phosphatase (ALP) activity was evaluated as a marker of osteogenic differentiation and function. To this end, primary osteoblasts were differentiated for 7 days, starved overnight (DMEM, 1% FCS, 1% P/S), and treated over 48 hours with indicated concentrations of T3, LDN193189, or noggin. After washing steps with PBS, cells were scraped in ALP lysis buffer (10 mM TrisHCl pH 8.0, 1 mM MgCl2, and 0.5% Triton X-100) for isolation of total proteins. Subsequently, cell lysates were processed through a 24-gauge needle and centrifuged at 25,000g for 30 minutes at 4C. Ten microliters of diluted supernatants (1:5 in distilled
water) were mixed with 90 μL ALP substrate buffer (100 mM diethanolamine, 150 mM NaCl, 2 mM MgCl2, and 2.5 g/mL p-nitrophenylphosphate) and incubated for 30 minutes at 37C. Color change was observed given the hydrolysis of p-nitrophenylphosphate by enzymatic activity of ALP. Absorbance was assessed at 410 nm using FluoStar Omega (BMG Labtech, Offenburg, Germany). Results were normalized to the total
protein content quantified with Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Schwerte, Germany) according to the manufacturer’s protocol. RNA isolation, reverse transcription, and quantitative real-
time PCR. After 7 days of differentiation, primary osteoblasts were starved
overnight and then treated with indicated concentrations of T3, LDN193189, noggin, or recombinant anti-BMP2 or anti-BMP4 neutralizing antibodies for 48 hours (Supplemental Table S2).
Total RNA was extracted with the High Pure RNA Isolation Kit
(Roche, Mannheim, Germany) according to the manufacturer’s
protocol and quantified using the Nanodrop spectrophotometer
(Peqlab, Erlangen, Germany). RNA (500 ng) was used for reverse transcription using Superscript II (Invitrogen, Darmstadt, Germany) and subsequently analyzed by SYBR green-based real-time PCRs using standard protocol (Applied Biosystems Inc., Carlsbad, CA, USA). PCR conditions were: 50C for 5 minutes and 95C for 10 minutes followed by 40 cycles with 95C for 15 seconds and 60C for 1 minute. Primer sequences for mice are listed in Supplemental Table S1. Melting curves were obtained by the following scheme: 95C for 15 seconds, 60C for 1 minute, and 95C for 30 seconds. Results were calculated according to the ΔΔCT method and are shown in x-fold increase normalized to β-actin mRNA levels.
Western blot analysis
Primary osteoblasts were differentiated over 7 days, starved overnight, and treated with 100 nM T3 for 20 minutes, 40 minutes, and 24 hours. With regard to inhibitor studies, cells were treated with 100 nM T3 and additionally 100 nM LDN193189 over 40 minutes. Subsequently, cells were lysed in lysis buffer containing 20 mM Tris/HCl pH 7.4, 1% SDS, a prote-
ase inhibitor cocktail (complete mini, Roche), and a phosphatase inhibitor cocktail (PhosSTOP, Roche). Samples were centrifuged at 25,000g at 4C for 20 minutes and total protein from supernatants was quantified using the BCA method. For electrophoresis, 20 μg protein was loaded on a 10% SDS-PAGE and subsequently transferred onto a 0.2 μm nitrocellulose membrane (Whatman). After 1 hour of blocking in 5% BSA in Tris-buffered saline with 1% Tween-20 (TBS-T), membranes were incubated overnight at 4C with primary antibodies listed in Supplemental Table S3. GAPDH was used as a loading control. Membranes were washed and incubated with the appropriate anti-rabbit or anti-mouse HRP conjugated secondary antibodies as listed in Supplemental Table S3. After washing and incubating the membranes with an ECL substrate (Pierce, Thermo Fisher Scientific), protein bands were visualized by MF-ChemiBIS 3.2 bioimaging system (Biostep, Jahnsdorf, Germany). The densitometric analysis of
the signal intensities was quantified using ImageJ 1.44 and normalized to GAPDH as the housekeeping protein and the corresponding unphosphorylated form of the protein of interest.
Statistical analysis
Data are shown as means standard deviation. Statistical analysis comparing two groups are based on a two-sided Student’s t test. One-way analysis of variance (ANOVA) or two-way ANOVA were performed for in vitro and in vivo experiments with more than two groups followed by Bonferroni’s multiple comparison post hoc test using GraphPad Prism 7.0 (GraphPad, La Jolla, CA, USA). Values of p < 0.05 were considered statistically significant.
Results
T3 enhances osteoblast differentiation and activity and activates canonical BMP signaling Treatment of primary murine osteoblasts with T3 led to
enhanced expression of differentiation marker genes including alkaline phosphatase (Alp; 100 nM T3: 2.2-fold), osteocalcin(Ocn; 100 nM T3: 6.6-fold), and osterix (Osx; 100 nM T3: 2.7-fold) in a dose-dependent manner (Fig. 1A–C). Moreover, osteoblast function was increased with T3 as shown by elevated ALP activity (100 nM T3: 1.6-fold) and mineralization capacity (100 nM T3: 1.5-fold) (Fig. 1D–F). Further, prominent BMP target genes
Runt-related transcription factor 2 (Runx2; 100 nM T3: 2-fold) and Inhibitor of DNA binding 3 (Id3; 100 nM T3: 2-fold) were transcriptionally upregulated after 48 hours of T3 treatment (Fig. 2A–C). Western blot analysis revealed canonical BMP pathway activation by enhanced SMAD1/5 phosphorylation after 40 minutes stimulation with 100 nM T3 indicating an immediate response at the protein level (Fig. 2D). SMAD-independent BMP
signaling was not activated as demonstrated by unaltered (pERK1/2; pP38) or even reduced phosphorylation (pAKT) of respective enzymes (Supplemental Fig. S1). Subsequently, mRNA levels of BMP target genes were assessed in T3-treated osteoblasts with regard to different treatment durations (1, 3, 6, 24 hours) uncovering peak expressions of Id2 and Id3 after 1 hour (2.5-fold/2.6-fold) (Fig. 2E–G). In conclusion, T3 directly
activates canonical BMP signaling via SMAD1/5 phosphorylation
with immediate transcriptional outcome. Blockade of BMP signaling by LDN193189 reverses T3- mediated effects in osteoblasts. To investigate whether T3-promoted osteoblastogenesis is mediated by BMP signaling, we inhibited its activation by specifically blocking BMPR type I members ALK2 and ALK3 using the small molecule LDN193189.(38,39) First, we confirmed signaling inhibition in T3-treated murine osteoblasts with LDN193189 at the protein level as indicated by a reduced SMAD1/5 phosphorylation status (Fig. 3A). Also, T3-induced expression of BMP target Runx2 as well as of osteogenic markers Ocn and Osx was downregulated after BMP blockade (Fig. 3B–D). In line with findings of
Kamiya and colleagues on Wnt inhibitor dickkopf-1 (Dkk1) expression being positively regulated by BMP signaling,(40) Dkk1 was upregulated by T3 and subsequently downregulated by adding LDN193189 (Fig. 3E). T3-mediated repression of Wnt target genes Axin2 and osteoprotegerin (Tnfrsf11b, internal abbreviation Opg), however, remained unchanged by
LDN193189 treatment (Fig. S2A, B). ALP activity and mineralization capacity of T3-stimulated osteoblasts were normalized through ALK2/ALK3 inhibition (Fig. 3F, G). As osteoblasts can regulate osteoclastogenesis by secretion of receptor activator of NF-κB ligand (RANKL) versus OPG, we further assessed the Rankl/Opg ratio. T3 treatment of osteoblasts resulted in an increased Rankl/Opg ratio likely benefiting osteoclast formation,
while LDN193189 normalized the ratio (Fig. 3H and Supplemental Fig. S2C).
BMP ligand scavenger noggin mitigates T3-induced osteogenic response
BMP signaling is activated upon BMP ligand binding to respective receptors. So far, BMP2, BMP4, BMP6, BMP7, and BMP9 were described as osteoinductive BMP ligands favoring commitment of MSC to the osteoblast lineage and further stimulating osteoblast differentiation and bone formation.(23–25) T3 increased expression of Bmp2 (100 nM T3: 1.7-fold) and Bmp4 (100 nM T3: 1.5-fold), whereas it reduced Bmp6 mRNA levels (100 nM T3:−46.2%) ex vivo (Fig. 4A–C). Expression of Bmp7 and Bmp9 was below the real-time PCR detection range. To test whether BMP ligands are crucially involved in T3-induced osteoblast activity, we used noggin, which sequesters specifically BMP2 and BMP4. Indeed, noggin normalized T3-activated expression of osteoblastic genes and osteoblast function (Fig. 4D–I). In addition, treatment of hyperthyroid osteoblasts with either specific antiBMP2 or anti-BMP4 neutralizing antibodies reversed T3-induced
expression of osteoblast marker genes (Supplemental Fig. Supplemental Fig. S3A, B) and BMP target genes (Supplemental Fig. Supplemental Fig. S3C, D). Thus, BMP signaling blockade at the receptor but also extracellular ligand level reversed T3-mediated effects on osteoblast differentiation and activity. BMP ligand trap ALK3-Fc protects against
hyperthyroidism-induced bone loss in male mice Hyperthyroidism leads to high bone turnover–induced bone loss due to predominant bone resorption.(9,21) Thus, we tested whether blockade of BMP signaling by scavenging BMP ligands may counteract the accelerated bone turnover and subsequent bone loss. To that end, we treated male hyperthyroid mice with ALK3-Fc, which was reported to sequester ALK3-targeting ligands such as BMP2 and BMP4. Treatment with ALK3-Fc was well-tolerated and no adverse effects were detected. Efficient induction of hyperthyroidism was shown by significant increases of total T3 and T4 serum concentrations in male T4-treated animals (Supplemental Fig. Supplemental Fig. S4). First, using micro-CT measurements, we confirmed trabecular bone loss at the L4 vertebra (−42.9%) and femora (−48.9%) of male mice caused by TH excess (Fig. 5A–J), while cortical bone volume
remained unchanged (Fig. 5K, L). Bone changes due to hyperthyroidism were observed in female mice as well (Supplemental Fig. S5). Moreover, bone microstructure analysis revealed a higher bone mass of male hyperthyroid mice treated with ALK3-Fc at the L4 and femur (2.1-fold/2.4-fold versus PBS-treated hyperthyroid mice) and an elevated trabecular number (1.2-fold/1.4-fold versus PBS-treated hyperthyroid mice), whereas trabecular separation was reduced (−16.4%/−28.2% versus PBS-treated hyperthyroid mice), respectively (Fig. 5A–J). ALK3-Fc treatment of euthyroid mice enhanced bone volume (1.4-fold/1.8-fold) and trabecular thickness (1.4-fold/1.4-fold), whereas trabecular number and separation were not altered (Fig. 5A–C). With regard to bone biomechanics, we observed low bone strength in hyperthyroid mice, which was improved
with ALK3-Fc (Fig. 5M). To assess bone formation, histological and bone histomorphometric analyses of vertebral bone slides were performed,
demonstrating augmented osteoblast numbers (1.8-fold), bone formation rate (2.9-fold), and mineral apposition rate (2.7-fold) in hyperthyroid mice (Fig. 6A–D). Inhibition of BMP signaling reversed the enhancement of bone formation parameters as well as the increase in P1NP serum concentrations (Fig. 6A–E). With regard to bone resorption, elevated osteoclast numbers due to hyperthyroidism (1.5-fold) were reduced by ALK3-Fc treatment (−48.3% versus PBS-treated, hyperthyroid mice) at the spine (Fig. 6F). ALK3-Fc also reduced osteoclast numbers in euthyroid
mice (−47.5% versus PBS-treated, euthyroid mice) (Fig. 6F, H). Further, serum concentration of the bone resorption marker TRAP was decreased in hyperthyroid mice with ALK3-Fc treatment (Fig. 6G). In summary, BMP ligand sequestration by ALK3-Fc protected against hyperthyroidism-induced bone loss likely due to normalization of bone formation as well as bone resorption.
Discussion
Bone homeostasis and skeletal health rely on a balanced TH status. In fact, TH excess in mice and humans leads to high bone turnover state with subsequent bone loss due to predominant bone resorption.(6–9,21) Hence, bone-forming osteoblasts as well as bone-resorbing osteoclasts are targeted by TH.(3,41–45) Similar to previous studies,(10–14) our results demonstrate that T3 augments osteoblast differentiation and function in vitro and in vivo. Moreover, we show that the pro-osteogenic effects of
T3 are mediated via activating canonical BMP signaling.
Our study reveals that T3 activates canonical but not noncanonical BMP signaling via rapid and more prolonged effects.
In line with in vitro studies that used BMP7, BMP4, or BMP2,(46–48) T3 induced BMP target gene expression within 1 hour. How this is achieved mechanistically is currently not known, but different scenarios can be envisaged. Considering that BMP must first bind to the BMPR and induce downstream SMAD signaling, the immediate effects of T3 on Id gene expression may indicate cooperative effects of SMADs with either liganded or unliganded TR directly in the nucleus. In fact, Alonso-Merino and colleagues reported on unoccupied TRs associating with SMAD4 and SMAD3 to enhance TGF-β signaling by increasing the activity or stability of transcriptional complexes. Interestingly, T3-binding obstructs this interaction, thereby inhibiting signaling transduction.(49) Finally, they showed that both TRα or TRβ were mediating these effects.(49) Whether TRα or TRβ are involved in BMP signaling in bone remains to be identified in future studies. Besides its effects on transcription, T3 enhanced SMAD1/5 phosphorylation after 40 minutes but not non-canonical SMAD-independent pathways. However, T3 was shown to induce PI3K-AKT signaling after 2 to 5 minutes but not 20 to 30 minutes in a previous study,(50) indicating specific time frames of distinct pathway activation. Also, p38 and ERK1/2-mediated signaling was activated with T3 in MC3T3-E1 cells after 1 hour and 3 hours stimulating osteocalcin synthesis and ALP activity, respectively.(51,52) Additional investigations should focus on time-dependent effects of T3 on SMAD dependent versus SMAD-independent pathway activation to resolve these discrepancies.
At the extracellular level, BMP signaling is tightly regulated via numerous ligands, agonists as well as antagonists. The mRNA levels of BMP ligands Bmp2 and Bmp4 but not Bmp6 were elevated with T3 after 48 hours. Noggin is a key inhibitor of BMP signaling by sequestering specifically BMP2 and BMP4 ligands.
Interestingly, noggin overexpression(53) and osteoblast-specific noggin inactivation(54) both lead to osteopenia, characterized by low bone mineral density and reduced bone formation. These studies suggest that either high BMP signaling activation can exhibit adverse effects on bone mass or that noggin can act BMP-independently on bone. Subsequently, anti-BMP2 and anti-BMP4 antibodies were used to selectively block these
ligands. Interestingly, both treatments were sufficient to abrogate T3-enhanced BMP signaling activation and osteoblast activity. Thus, T3-mediated induction of BMP2 and BMP4 may represent an autocrine feed-forward loop that is involved in the pro-osteogenic effects of T3. However, not only sequestering BMP ligands but also blocking BMPR type I-mediated signaling transduction with LDN193189 reversed the positive effects of
T3 on osteoblast differentiation. T3-dependent effects on bone were also mitigated by blocking BMP signaling in vivo. Using our established protocol, we are able to induce hyperthyroidism-induced bone loss in both
male and female mice. However, we included only male mice
in this initial mechanistic investigation, because several studies
implicated a close cross-talk between BMP and estrogen recep-
tor signaling in osteoblasts/bone that could introduce unex-
pected variables in our experiments.(55,56) To test whether BMP signaling activation mediates thyroid hormone actions in bone, we treated male mice with the BMP antagonist ALK3-Fc that has been previously shown to increase bone mass in healthy mice and to prevent estrogen deficiency–induced osteoporosis.(57) Mechanistically, ALK3-Fc promotes bone formation and reduces bone resorption.(57) In our study, however, ALK3-Fc mitigated hyperthyroidism-induced bone loss by normalizing bone turnover, ie, reducing bone formation and bone resorption parameters to control levels. This is in line with studies on Alk3 knockout mice, which show increased bone mass with low bone turnover, suggesting that the reduction of bone resorption is more severe than the reduction of bone formation.(58–61) The suppression of osteoclastogenesis in mice lacking Alk3 specifically in osteoblasts or osteocytes seems to be mediated via a low RANKL/OPG ratio in Alk3 deficient osteogenic cells.(58) Similarly, we demonstrated a normalization of the Rankl/Opg ratio by BMP inhibition ex vivo that might contribute to reduced osteoclast numbers. In addition,
osteoclasts might be directly influenced by BMP signaling. Genetic and pharmacological blockade of BMP signaling has been shown to affect osteoclasts in vitro and in vivo, especially restricting osteoclastic formation and function.(38,62,63) Further, exogenously applied BMP2 can promote RANKL-mediated osteoclastogenesis and osteoclast activity, implying that BMP locally secreted by osteoblasts might trigger those effects in a
similar manner.(64) Vice versa, BMP signaling in osteoclasts also appears to alter osteoblast function as inhibited SMAD1/5 signaling in osteoclasts has been shown to increase osteoblast activity via osteoclast-to-osteoblast coupling factors such as WNT1, gap junction alpha-1 protein, and sphingosine kinase 1.(65)
Finally, recent reports indicate tight connections between BMP and Wnt signaling.(40,66) in vitro blockade of the BMP pathway reversed T3-stimulated Dkk1 expression but did not alter the expression of Wnt target genes Axin2 nor Opg, suggesting additional mechanisms might obstruct Wnt signaling in osteoblasts.
Previous studies demonstrated that unliganded TRβ binds and stabilizes β-catenin, while T3-binding dissociates their physical connection, leading to β-catenin degradation and subsequent inhibition of canonical Wnt signaling.(67,68) Therefore, canonical Wnt signaling might be regulated independently from BMP signaling in hyperthyroid mice.
Taken together, our study revealed that T3 augments osteoblastogenesis and osteoblast activity via canonical BMP signaling. Moreover, blocking BMP signaling may be an effective strategy to treat hyperthyroidism-induced bone loss.
Disclosures
MR reports honoraria for lectures from Amgen. LCH reports honoraria for advisory boards from Alexion, Amgen, Merck, Radius, Roche, Shire, and UCB to his institution and himself. ET reports honoraria for lectures from Amgen and educational grants from UCB and Shire. RK is employed by Acceleron Pharma, Inc. JK reports honoraria for lectures from Berlin-Chemie, Ipsen, and Sanofi/Genzyme to his institution and himself. The remaining authors have nothing to disclose.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft Schwerpunktprogramm 1629 ThyroidTransAct (to LCH and MR). We thank Ina Gloe, Tina Dybek, Maria Heier, Sandra Hippauf, and Eva Schubert for excellent technical assistance. Authors’ roles: FL and MR designed the experiments. FL, HW, ET, and MR performed all mouse and in vitro experiments and analyzed the data. All authors contributed to data discussion and interpretation. FL and MR drafted the manuscript. All authors critically read, revised, and approved the final version of the
manuscript.
References
1. Crockett JC, Rogers MJ, Coxon FP. Bone remodelling at a glance. J Cell
Sci. 2011;124:991–8.
2. Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone
remodeling. J Biol Chem. 2010;285:25103–8.
3. Bassett JHD, Williams GR. Role of thyroid hormones in skeletal devel-
opment and bone maintenance. Endocr Rev. 2016;37:135–87.
4. Waung JA, Bassett JHD, Williams GR. Thyroid hormone metabolism in
skeletal development and adult bone maintenance. Trends Endocri-
nol Metab. 2012;23:155–62.
5. Lazarus JH, Obuobie K. Thyroid disorders-an update. Postgrad Med J.
2000;76:529–36.
6. Vestergaard P, Mosekilde L. Hyperthyroidism, bone mineral, and frac-
ture risk—a meta-analysis. Thyroid. 2003;13:585–93.
7. Vestergaard P, Rejnmark L, Mosekilde L. Influence of hyper- and
hypothyroidism, and the effects of treatment with antithyroid drugs
and levothyroxine on fracture risk. Calcif Tissue Int. 2005;77:139–44.
8. Nicholls JJ, Brassill MJ, Williams GR. The skeletal consequences of thy-
rotoxicosis. J Endocrinol. 2012;213:209–21.
9. Tsourdi E, Rijntjes E, Köhrle J. Hyperthyroidism and hypothyroidism in
male mice and their effects on bone mass, bone turnover, and the
Wnt inhibitors sclerostin and dickkopf-1. Endocrinology. 2015;156:
3517–27.
10. Fratzl-Zelman N, Hörandner H, Luegmayr E. Effects of triiodothyro-
nine on the morphology of cells and matrix, the localization of alka-
line phosphatase, and the frequency of apoptosis in long-term
cultures of MC3T3-E1 cells. Bone. 1997;20:225–36.
11. Klaushofer K, Varga F, Glantschnig H. The regulatory role of thyroid
hormones in bone cell growth and differentiation. J Nutr. 2018;125:
1996S–2003S.
12. Varga F, Rumpler M, Zoehrer R. T3 affects expression of collagen I and
collagen cross-linking in bone cell cultures. Biochem Biophys Res
Commun. 2010;402:180–5.
13. Banovac K, Koren E. Triiodothyronine stimulates the release of
membrane-bound alkaline phosphatase in osteoblastic cells. Calcif
Tissue Int. 2000;67:460–5.
14. Tokuda K, Otsuka T, Adachi S. (−)-epigallocatechin gallate inhibits
thyroid hormone-stimulated osteocalcin synthesis in osteoblasts.
Mol Med Rep. 2011;4:297–300.
15. Cray JJ, Khaksarfard K, Weinberg SM. Effects of thyroxine exposure on
osteogenesis in mouse Calvarial pre-osteoblasts. PLoS One. 2013;8:
e69067.
16. Huang BK, Golden LA, Tarjan G. Insulin-like growth factor I produc-
tion is essential for anabolic effects of thyroid hormone in osteo-
blasts. J Bone Miner Res. 2010;15:188–97.
17. Siddiqi A, Parsons MP, Lewis JL. TR expression and function in human
bone marrow stromal and osteoblast-like cells. J Clin Endocrinol
Metab. 2002;87:906–14.
18. Gruber R, Czerwenka K, Wolf F. Expression of the vitamin D receptor,
of estrogen and thyroid hormone receptor alpha- and beta-isoforms,
and of the androgen receptor in cultures of native mouse bone mar-
row and of stromal/osteoblastic cells. Bone. 1999;24:465–73.
19. Allain TJ, Yen PM, Flanagan AM. The isoform-specific expression of
the tri-iodothyronine receptor in osteoblasts and osteoclasts. Eur J
Clin Invest. 1996;26:418–25.
20. Krieger NS, Stappenbeck TS, Stern PH. Characterization of specific
thyroid hormone receptors in bone. J Bone Miner Res. 1988;3:473–8.
21. Tsourdi E, Lademann F, Ominsky MS. Sclerostin blockade and zole-
dronic acid improve bone mass and strength in male mice with exog-
enous hyperthyroidism. Endocrinology. 2017;158:3765–77.
22. Tsourdi E, Colditz J, Lademann F. The role of Dickkopf-1 in thyroid
hormone-induced changes of bone remodeling in male mice. Endo-
crinology. 2019;160:664–74.
23. Rahman MS, Akhtar N, Jamil HM. TGF-β/BMP signaling and other
molecular events: regulation of osteoblastogenesis and bone forma-
tion. Bone Res. 2015;3:15005.
24. Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein
receptors and signal transduction. J Biochem. 2010;147:35–51.
25. Wu M, Chen G, Li YP. TGF-β and BMP signaling in osteoblast, skeletal
development, and bone formation, homeostasis and disease. Bone
Res. 2016;4:16009.
26. Beederman M, Lamplot JD, Nan G. BMP signaling in mesenchymal
stem cell differentiation and bone formation. J Biomed Sci Eng.
2013;6:32–52.
27. Massague J, Seoane J, Wotton D. Smad transcription factors. Genes
Dev. 2005;19:2783–810.
28. Greenblatt MB, Shim J-H, Zou W. The p38 MAPK pathway is essential
for skeletogenesis and bone homeostasis in mice. J Clin Invest. 2010;
120:2457–73.
29. Ortuño MJ, Ruiz-Gaspà S, Rodríguez-Carballo E. p38 regulates expres-
sion of osteoblast-specific genes by phosphorylation of Osterix. J Biol
Chem. 2010;285:31985–94.
30. Choi YH, Gu Y-M, Oh J-W. Osterix is regulated by Erk1/2 during oste-
oblast differentiation. Biochem Biophys Res Commun. 2011;415:472–8.
31. Ge C, Yang Q, Zhao G. Interactions between extracellular signal-
regulated kinase 1/2 and p38 MAP kinase pathways in the control
of RUNX2 phosphorylation and transcriptional activity. J Bone Miner
Res. 2012;27:538–51.
32. Lauzon M-A, Drevelle O, Daviau A. Effects of BMP-9 and BMP-2 on the
PI3K/Akt pathway in MC3T3-E1 preosteoblasts. Tissue Eng Part A.
2016;22:1075–85.
33. Lassová L, Niu Z, Golden EB. Thyroid hormone treatment of cultured
chondrocytes mimics in vivo stimulation of collagen X mRNA by
increasing BMP 4 expression. J Cell Physiol. 2009;219:595–605.
34. Bouxsein ML, Boyd SK, Christiansen BA. Guidelines for assessment of
bone microstructure in rodents using micro-computed tomography.
J Bone Miner Res. 2010;25:1468–86.
35. Rauner M, Föger-Samwald U, Kurz MF. Cathepsin S controls adipocy-
tic and osteoblastic differentiation, bone turnover, and bone micro-
architecture. Bone. 2014;64:281–7.
36. Rauner M, Franke K, Murray M. Increased EPO levels are associated
with bone loss in mice lacking PHD2 in EPO-producing cells. J Bone
Miner Res. 2016;31:1877–87.
37. Dempster DW, Compston JE, Drezner MK. Standardized nomencla-
ture, symbols, and units for bone histomorphometry: a 2012 update
of the report of the ASBMR Histomorphometry Nomenclature Com-
mittee. J Bone Miner Res. 2013;28:2–17.
38. Boergermann JH, Kopf J, Yu PB. Dorsomorphin and LDN-193189
inhibit BMP-mediated Smad, p38 and Akt signalling in C2C12 cells.
Int J Biochem Cell Biol. 2010;42:1802–7.
39. Yu PB, Deng DY, Lai CS. BMP type I receptor inhibition reduces het-
erotopic ossification. Nat Med. 2008;14:1363–9.
40. Kamiya N, Kobayashi T, Mochida Y. Wnt inhibitors Dkk1 and sost are
downstream targets of BMP signaling through the type IA receptor
(BMPRIA) in osteoblasts. J Bone Miner Res. 2010;25:200–10.
41. Klaushofer K, Hoffmann O, Gleispach H. Bone-resorbing activity of
thyroid hormones is related to prostaglandin production in cultured
neonatal mouse calvaria. J Bone Miner Res. 2009;4:305–12.
42. Mundy GR, Shapiro JL, Bandelin JG. Direct stimulation of bone
resorption by thyroid hormones. J Clin Invest. 1976;58:529–34.
43. Allain TJ, Chambers TJ, Flanagan AM. Tri-iodothyronine stimulates rat
osteoclastic bone resorption by an indirect effect. J Endocrinol. 1992;
133:327–31.
44. Siddiqi A, Burrin JM, Wood DF. Tri-iodothyronine regulates the pro-
duction of interleukin-6 and interleukin-8 in human bone marrow
stromal and osteoblast-like cells. J Endocrinol. 1998;157:453–61.
45. Miura M, Tanaka K, Komatsu Y. A novel interaction between thyroid
hormones and 1,25(OH)2D3 in osteoclast formation. Biochem Bio-
phys Res Commun. 2002;291:987–94.
46. Korchynskyi O, ten Dijke P. Identification and functional characteriza-
tion of distinct critically important bone morphogenetic protein-
specific response elements in the Id1 promoter. J Biol Chem. 2002;
277:4883–91.
47. Hollnagel A, Oehlmann V, Heymer J. Id genes are direct targets of
bone morphogenetic protein induction in embryonic stem cells.
J Biol Chem. 1999;274:19838–45.
48. López-Rovira T, Chalaux E, Massagué J. Direct binding of Smad1 and
Smad4 to two distinct motifs mediates bone morphogenetic protein-
specific transcriptional activation of Id1 gene. J Biol Chem. 2002;277:
3176–85.
49. Alonso-Merino E, Martín Orozco R, Ruíz-Llorente L. Thyroid hormones
inhibit TGF-β signaling and attenuate fibrotic responses. Proc Natl
Acad Sci. 2016;113:E3451–60.
50. Kalyanaraman H, Schwappacher R, Joshua J. Nongenomic thyroid
hormone signaling occurs through a plasma membrane -localized
receptor. Sci Signal. 2014;7:ra48.
51. Ishisaki A, Tokuda H, Yoshida M. Activation of p38 mitogen-activated
protein kinase mediates thyroid hormone-stimulated osteocalcin
synthesis in osteoblasts. Mol Cell Endocrinol. 2004;214:189–95.
52. Kozawa O, Hatakeyama D, Yoshida M. Activation of p44/p42
mitogen-activated protein kinase limits triiodothyronine-stimulated
alkaline phosphatase activity in osteoblasts. Biochem Biophys Res
Commun. 2001;286:1140–3.
53. Devlin RD, Du Z, Pereira RC. Skeletal overexpression of noggin results
in osteopenia and reduced bone formation. Endocrinology. 2003;
144:1972–8.
54. Canalis E, Brunet LJ, Parker K. Conditional inactivation of noggin in
the postnatal skeleton causes osteopenia. Endocrinology. 2012;153:
1616–26.
55. Soldin OP, Mattison DR. Sex differences in pharmacokinetics and
pharmacodynamics. Clin Pharmacokinet. 2009;48:143–57.
56. Salama G, Bett GCL. Sex and gender differences in cardiovascular
physiology–back to basics: sex differences in the mechanisms under-
lying long QT syndrome. Am J Physiol. 2014;307:H640.
57. Baud’huin M, Solban N, Cornwall-Brady M. A soluble bone morpho-
genetic protein type IA receptor increases bone mass and bone strength. Proc Natl Acad Sci. 2012;109:12207–12.
58. Kamiya N, Ye L, Kobayashi T. Disruption of BMP signaling in osteo-
blasts through type IA receptor (BMPRIA) increases bone mass.
J Bone Miner Res. 2008;23:2007–17.
59. Kamiya N, Shuxian L, Yamaguchi R. Targeted disruption of BMP sig-
naling through type IA receptor (BMPR1A) in osteocyte suppresses
SOST and RANKL, leading to dramatic increase in bone mass, bone
mineral density and mechanical strength. Bone. 2016;91:53–63.
60. Zhang Y, McNerny EG, Terajima M. Loss of BMP signaling through
BMPR1A in osteoblasts leads to greater collagen cross-link matura-
tion and material-level mechanical properties in mouse femoral trabecular compartments. Bone. 2016;88:74–84.
61. Shi C, Mandair GS, Zhang H. Bone morphogenetic protein signaling
through ACVR1 and BMPR1A negatively regulates bone mass along
with alterations in bone composition. J Struct Biol. 2018;201:237–46.
62. Okamoto M, Murai J, Imai Y. Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J Bone Miner Res. 2011;26:
2511–22.
63. Tasca A, Stemig M, Broege A. Smad1/5 and Smad4 expression are
important for osteoclast differentiation. J Cell Biochem. 2015;116:
1350–60.
64. Jensen ED, Pham L, Billington CJ. Bone morphogenic protein
2 directly enhances differentiation of murine osteoclast precursors.
J Cell Biochem. 2010;109:672–82.
65. Tasca A, Astleford K, Blixt NC. SMAD1/5 signaling in osteoclasts regu-
lates bone formation via coupling factors. PLoS One. 2018;13:
e0203404.
66. Rauner M, Baschant U, Roetto A. Transferrin receptor 2 controls bone
mass and pathological bone formation via BMP and Wnt signalling.
Nat Metab. 2018;1:111–24.
67. O’Shea PJ, Kim DW, Logan JG. Advanced bone formation in mice with
a dominant-negative mutation in the thyroid hormone receptor β
gene due to activation of Wnt/β-catenin protein signaling. J Biol
Chem. 2012;287:17812–22.
68. Guigon CJ, Zhao L, Lu C. Regulation of beta-catenin by a novel non-
genomic action of thyroid hormone receptor. Mol Cell Biol. 2008;
28:4598–608.



